
Structure and Bonding Patterns in C5H4 Isomers: Pyramidane, Planar Tetracoordinate Carbon, and Spiro Molecules

 ,
,  , ,
, ,  ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Computational Methodology
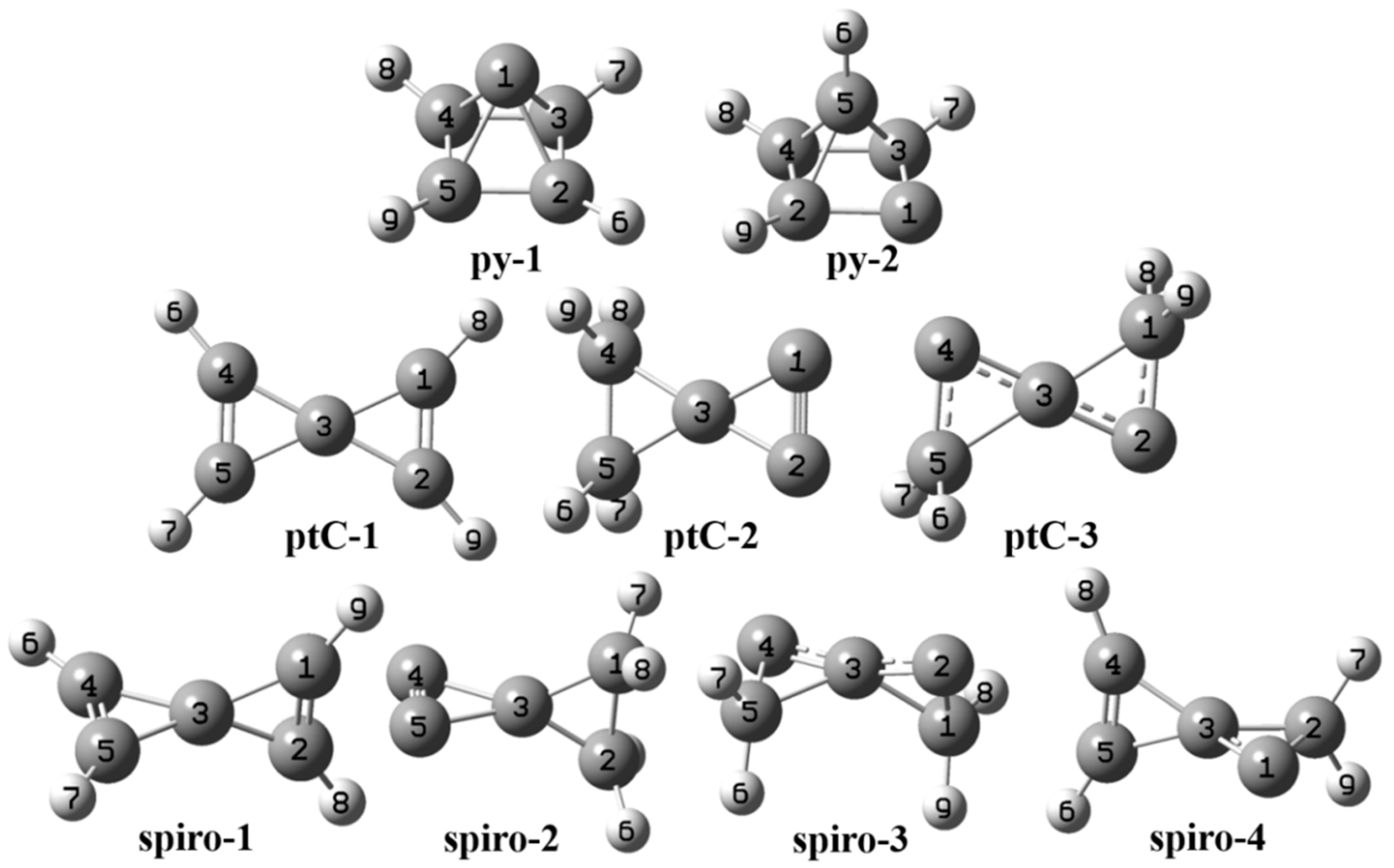
3. Results and Discussions
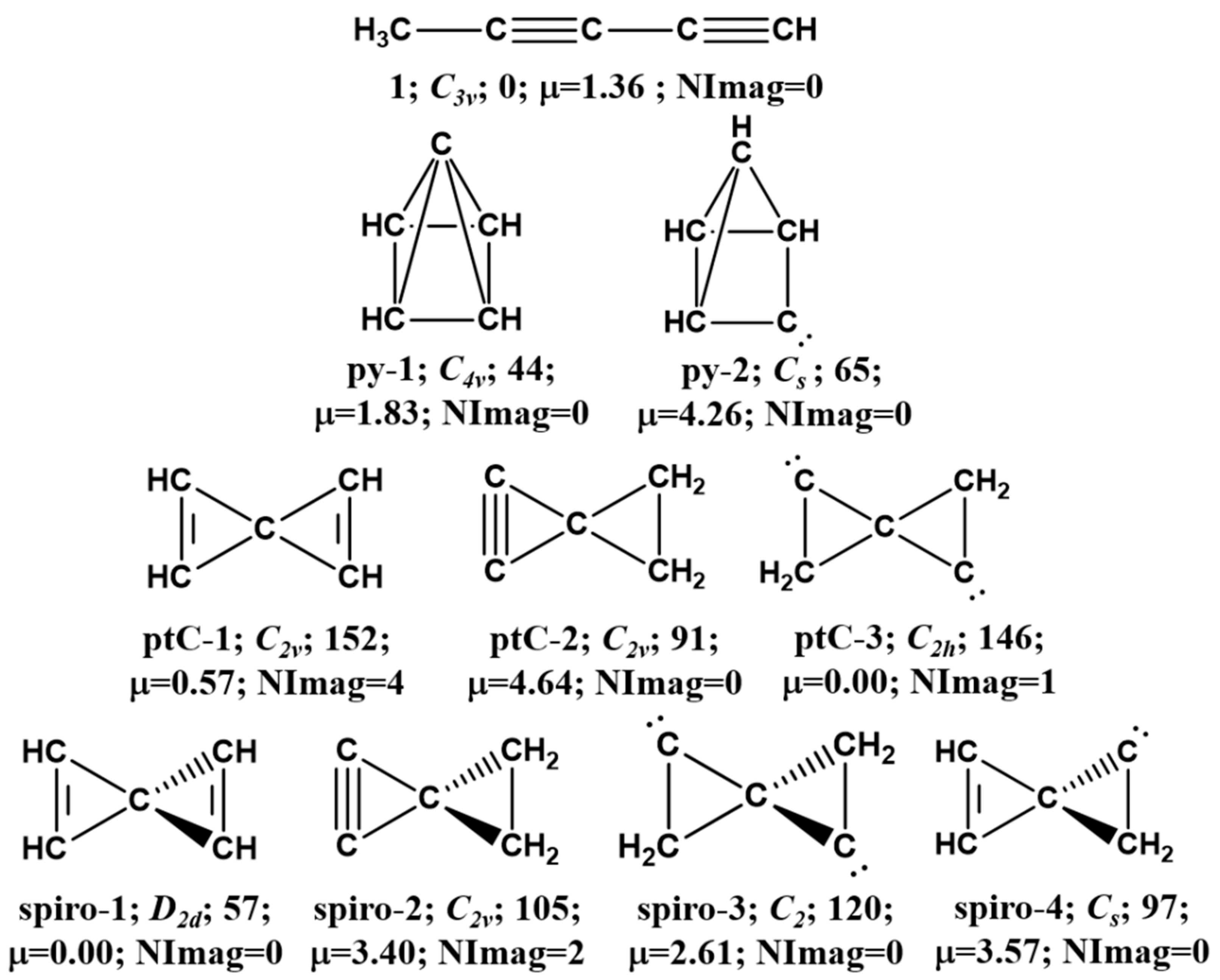
3.1. Pyramidane
3.2. Planar Tetracoordinate Carbon (ptC)
3.3. Spiro
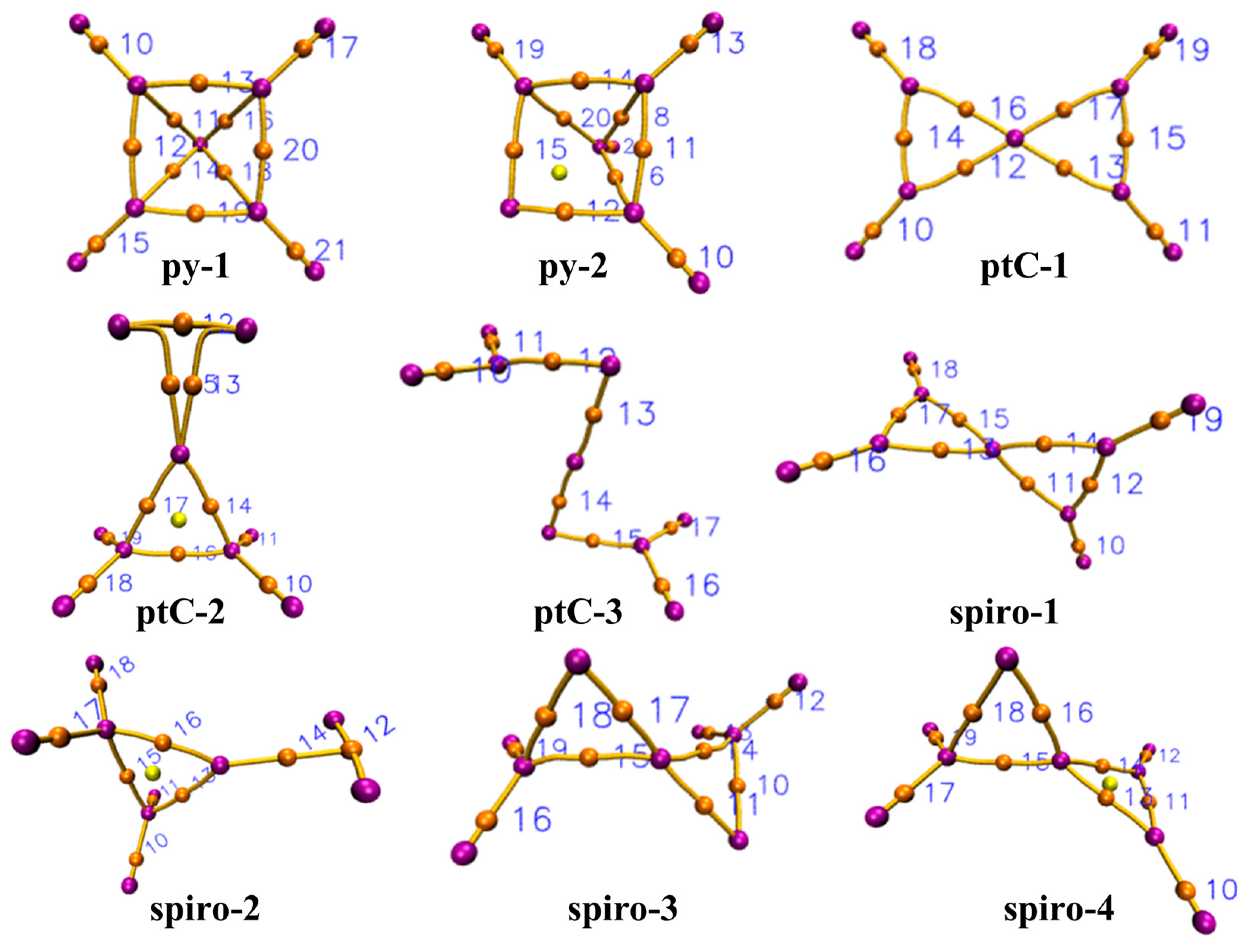
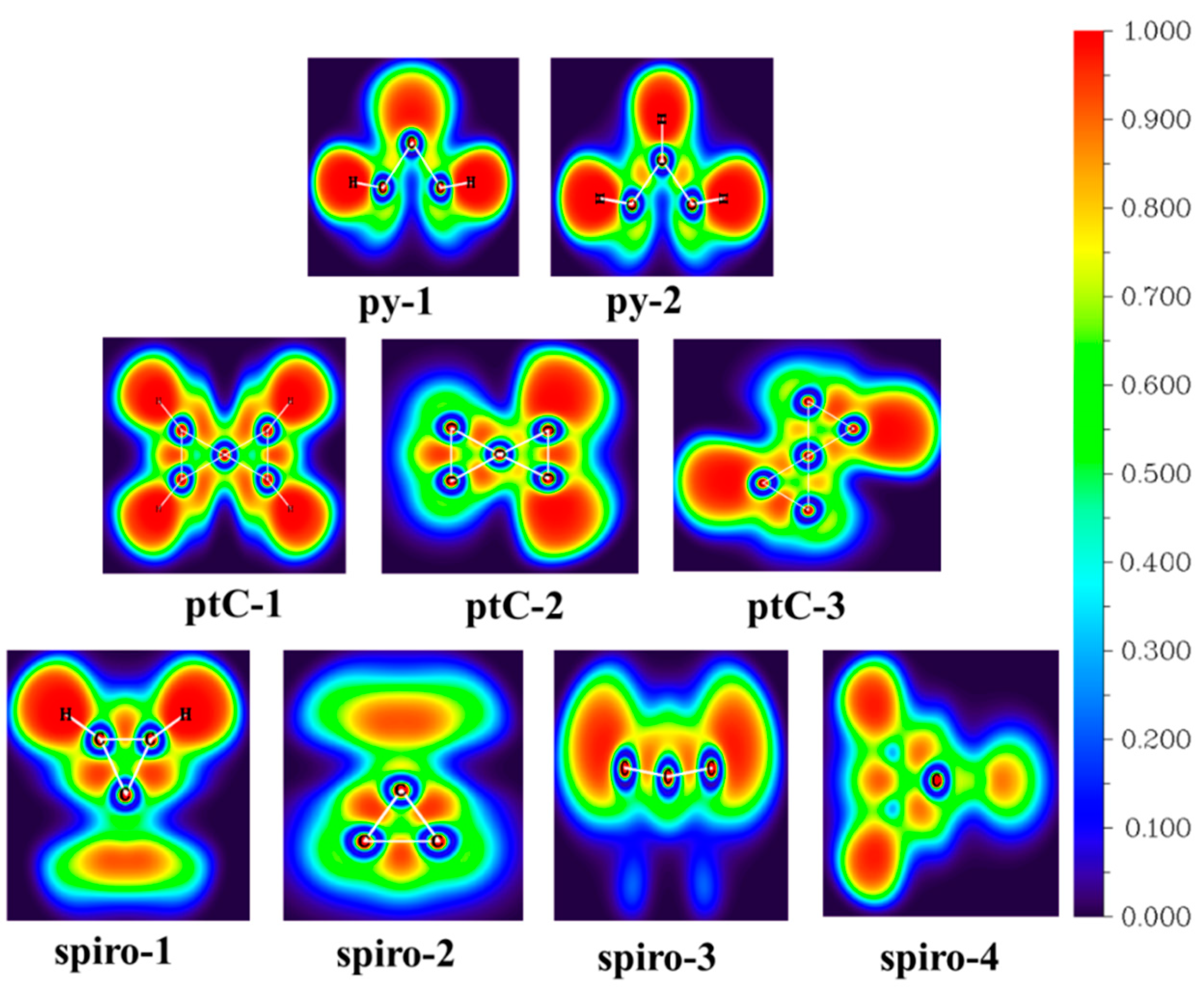
3.4. Topological Analysis using AIM Theory
3.5. Structural Comparison between Different Spin States
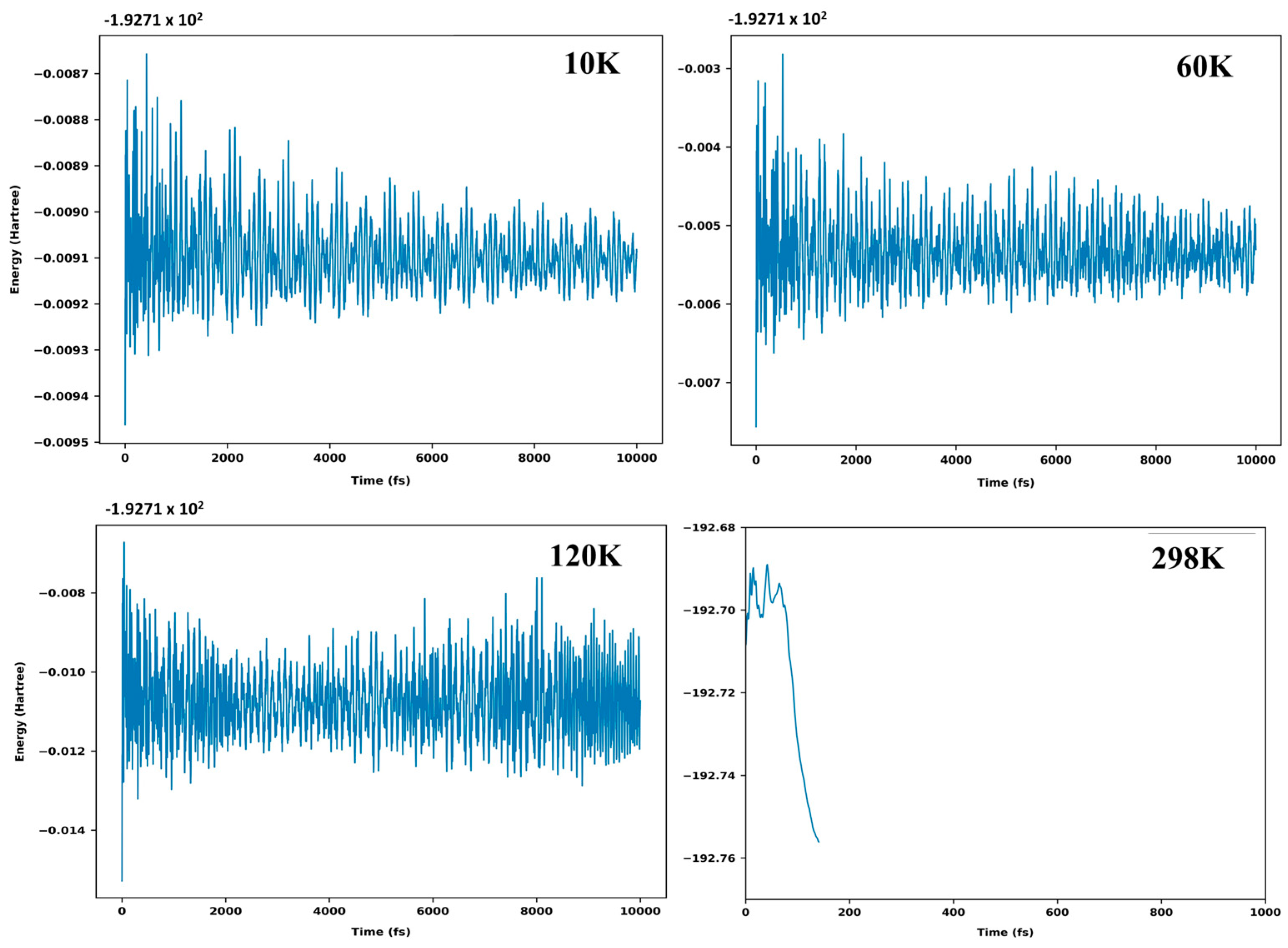
3.6. Ab Initio Molecular Dynamics
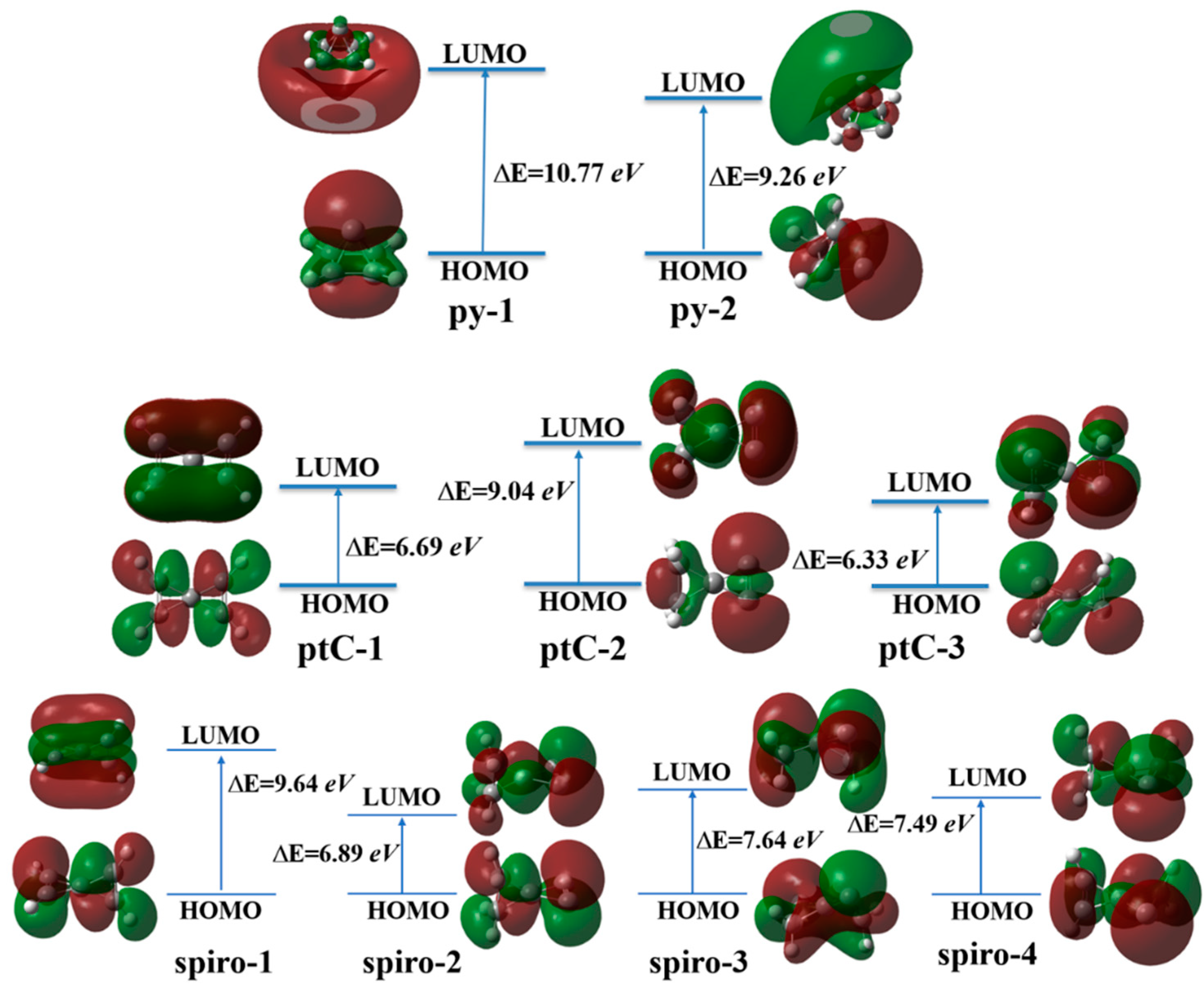
3.7. Aromatic Characteristics and Relative Stability of the Molecules
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van’t Hoff, J.H. A suggestion looking to the extension into space of the structural formulas at present used in chemistry, and a note upon the relation between the optical activity and the chemical constitution of organic compounds. Arch. Neerl. Sci. Exactes Nat. 1874, 9, 445–454. [Google Scholar]
- Le Bel, J.A. Sur les relations qui existent entre les formules atomiques des corps organiques et le pouvoir rotatoire de leurs dissolutions. Bull. Soc. Chim. Fr. 1874, 22, 337–347. [Google Scholar]
- Minkin, V.I.; Minyaev, R.M.; Hoffmann, R. Non-classical structures of organic compounds: Unusual stereochemistry and hypercoordination. Russ. Chem. Rev. 2002, 71, 869–892. [Google Scholar] [CrossRef]
- Vassilev-Galindo, V.; Pan, S.; Donald, K.J.; Merino, G. Planar pentacoordinate carbons. Nat. Rev. Chem. 2018, 2, 0114. [Google Scholar] [CrossRef]
- Yang, L.M.; Ganz, E.; Chen, Z.; Wang, Z.X.; Schleyer, P.V.R. Four decades of the chemistry of planar hypercoordinate compounds. Angew. Chem. Int. Ed. 2015, 54, 9468–9501. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Dong, S.; Yao, S.; Zhu, J.; Driess, M. Synthesis and Reactivity of an Anti-van’t Hoff/Le Bel Compound with a Planar Tetracoordinate Silicon(II) Atom. J. Am. Chem. Soc. 2023, 145, 7084–7089. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xia, D.; Baumgarten, M. Rigidly Fused Spiro-Conjugated π-Systems. ChemPlusChem 2021, 86, 36–48. [Google Scholar]
- Randić, M.; Rubčić, A.; Klasinc, L. Hybridization in highly strained small ring hydrocarbons—III: Unsaturated spiro-compounds. Tetrahedron 1971, 27, 5771–5777. [Google Scholar] [CrossRef]
- Delouche, T.; Hissler, M.; Bouit, P.-A. Polycyclic aromatic hydrocarbons containing heavy group 14 elements: From synthetic challenges to optoelectronic devices. Coord. Chem. Rev. 2022, 464, 214553. [Google Scholar] [CrossRef]
- Firme, C.L. Cycloalkanes, Bicyclic, and Caged Hydrocarbons. In Introductory Organic Chemistry and Hydrocarbons; CRC Press: Boca Raton, FL, USA, 2019; pp. 241–266. [Google Scholar]
- Leyva-Parra, L.; Inostroza, D.; Yañez, O.; Cruz, J.C.; Garza, J.; García, V.; Tiznado, W. Persistent planar tetracoordinate carbon in global minima structures of silicon-carbon clusters. Atoms 2022, 10, 27. [Google Scholar] [CrossRef]
- Pancharatna, P.D.; Méndez-Rojas, M.A.; Merino, G.; Vela, A.; Hoffmann, R. Planar tetracoordinate carbon in extended systems. J. Am. Chem. Soc. 2004, 126, 15309–15315. [Google Scholar] [CrossRef] [PubMed]
- Thirumoorthy, K.; Thimmakondu, V.S. Flat crown ethers with planar tetracoordinate carbon atoms. Int. J. Quantum. Chem. 2021, 121, e26479. [Google Scholar] [CrossRef]
- Thirumoorthy, K.; Karton, A.; Thimmakondu, V.S. From High-Energy C7H2 Isomers with A Planar Tetracoordinate Carbon Atom to An Experimentally Known Carbene. J. Phys. Chem. A 2018, 122, 9054–9064. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-S.; Boldyrev, A.I.; Li, X.; Simons, J. Experimental Observation of Pentaatomic Tetracoordinate Planar Carbon-Containing Molecules. J. Am. Chem. Soc. 2000, 122, 7681–7687. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Yu, S.; Ding, Y.-H.; Bowen, K.H. Identifying the Hydrogenated Planar Tetracoordinate Carbon: A Combined Experimental and Theoretical Study of CAl4H and CAl4H−. J. Phys. Chem. Lett. 2017, 8, 2263–2267. [Google Scholar] [CrossRef]
- Röttger, D.; Erker, G. Compounds Containing Planar-Tetracoordinate Carbon. Angew. Chem. Int. Ed. 1997, 36, 812–827. [Google Scholar] [CrossRef]
- Hoffmann, R.; Alder, R.W.; Wilcox, C.F., Jr. Planar tetracoordinate carbon. J. Am. Chem. Soc. 1970, 92, 4992–4993. [Google Scholar] [CrossRef]
- McGrath, M.P.; Radom, L. Alkaplanes: A Class of Neutral Hydrocarbons Containing a Potentially Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 1993, 115, 3320–3321. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Schleyer, P.v.R. The theoretical design of neutral planar tetracoordinate carbon molecules with C (C) 4 substructures. J. Am. Chem. Soc. 2002, 124, 11979–11982. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Schleyer, P.v.R. A new strategy to achieve perfectly planar carbon tetracoordination. J. Am. Chem. Soc. 2001, 123, 994–995. [Google Scholar] [CrossRef]
- Sorger, K.; Ragué; Schleyer, P.V. Planar and inherently non-tetrahedral tetracoordinate carbon: A status report. J. Mol. Struct. 1995, 338, 317–346. [Google Scholar] [CrossRef]
- Siebert, W.; Gunale, A. Compounds containing a planar-tetracoordinate carbon atom as analogues of planar methane. Chem. Soc. Rev. 1999, 28, 367–371. [Google Scholar] [CrossRef]
- Rasmussen, D.R.; Radom, L. Planar-Tetracoordinate Carbon in a Neutral Saturated Hydrocarbon: Theoretical Design and Characterization. Angew. Chem. Int. Ed. 1999, 38, 2875–2878. [Google Scholar] [CrossRef]
- Das, P.; Khatun, M.; Anoop, A.; Chattaraj, P.K. CSinGe4−n2+ (n = 1–3): Prospective systems containing planar tetracoordinate carbon (ptC). Phys. Chem. Chem. Phys. 2022, 24, 16701–16711. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.X.; Von Ragué Schleyer, P. Construction principles of "Hyparenes": Families of molecules with planar pentacoordinate carbons. Science 2001, 292, 2465–2469. [Google Scholar] [CrossRef]
- Yañez, O.; Vásquez-Espinal, A.; Báez-Grez, R.; Rabanal-León, W.A.; Osorio, E.; Ruiz, L.; Tiznado, W. Carbon rings decorated with group 14 elements: New aromatic clusters containing planar tetracoordinate carbon. N. J. Chem. 2019, 43, 6781–6785. [Google Scholar] [CrossRef]
- Wang, M.-H.; Orozco-Ic, M.; Leyva-Parra, L.; Tiznado, W.; Barroso, J.; Ding, Y.-H.; Cui, Z.-H.; Merino, G. Planar Tetracoordinate Carbons in Allene-Type Structures. J. Phys. Chem. A 2021, 125, 3009–3014. [Google Scholar] [CrossRef] [PubMed]
- Job, N.; Khatun, M.; Thirumoorthy, K.; CH, S.S.R.; Chandrasekaran, V.; Anoop, A.; Thimmakondu, V.S. CAl4Mg0/−: Global Minima with a Planar Tetracoordinate Carbon Atom. Atoms 2021, 9, 24. [Google Scholar] [CrossRef]
- Sateesh, B.; Srinivas Reddy, A.; Narahari Sastry, G. Towards design of the smallest planar tetracoordinate carbon and boron systems. J. Comput. Chem. 2007, 28, 335–343. [Google Scholar] [CrossRef]
- Suresh, C.H.; Frenking, G. Direct 1−3 Metal−Carbon Bonding and Planar Tetracoordinated Carbon in Group 6 Metallacyclobutadienes. Organometallics 2010, 29, 4766–4769. [Google Scholar] [CrossRef]
- Thirumoorthy, K.; Chandrasekaran, V.; Cooksy, A.L.; Thimmakondu, V.S. Kinetic Stability of Si2C5H2 Isomer with a Planar Tetracoordinate Carbon Atom. Chemistry 2020, 3, 13–27. [Google Scholar] [CrossRef]
- Rasmussen, D.R.; Radom, L. Hemispiroalkaplanes: Hydrocarbon Cage Systems with a Pyramidal-Tetracoordinate Carbon Atom and Remarkable Basicity. Chem. Eur. J. 2000, 6, 2470–2483. [Google Scholar] [CrossRef] [PubMed]
- Priyakumar, U.D.; Reddy, A.S.; Sastry, G.N. The design of molecules containing planar tetracoordinate carbon. Tetrahedron Lett. 2004, 45, 2495–2498. [Google Scholar] [CrossRef]
- Karton, A.; Thimmakondu, V.S. From Molecules with a Planar Tetracoordinate Carbon to an Astronomically Known C5H2 Carbene. J. Phys. Chem. A 2022, 126, 2561–2568. [Google Scholar] [CrossRef]
- Cabezas, C.; Tercero, B.; Agúndez, M.; Marcelino, N.; Pardo, J.; de Vicente, P.; Cernicharo, J. Cumulene carbenes in TMC-1: Astronomical discovery of l-H2C5. Astron. Astrophys. 2021, 650, L9. [Google Scholar] [CrossRef]
- Cernicharo, J.; Agúndez, M.; Cabezas, C.; Tercero, B.; Marcelino, N.; Pardo, J.R.; de Vicente, P. Pure hydrocarbon cycles in TMC-1: Discovery of ethynyl cyclopropenylidene, cyclopentadiene, and indene. Astron. Astrophys. 2021, 649, L15. [Google Scholar] [CrossRef]
- Blanksby, S.J.; Dua, S.; Bowie, J.H.; Schröder, D.; Schwarz, H. Gas-phase syntheses of three isomeric C5H2 radical anions and their elusive neutrals. a joint experimental and theoretical study. J. Phys. Chem. A 1998, 102, 9949–9956. [Google Scholar] [CrossRef]
- He, C.; Galimova, G.R.; Luo, Y.; Zhao, L.; Eckhardt, A.K.; Sun, R.; Mebel, A.M.; Kaiser, R.I. A chemical dynamics study on the gas-phase formation of triplet and singlet C5H2 carbenes. Proc. Natl. Acad. Sci. USA 2020, 117, 30142–30150. [Google Scholar] [CrossRef]
- Sun, Y.-L.; Huang, W.-J.; Lee, S.-H. Formation of C3H2, C5H2, C7H2, and C9H2 from reactions of CH, C3H, C5H, and C7H radicals with C2H2. Phys. Chem. Chem. Phys. 2016, 18, 2120–2129. [Google Scholar] [CrossRef]
- Jayakumari, C.M.; Nag, P.; Isukapalli, S.V.K.; Vennapusa, S.R. Exploring the Excited-State Nonadiabatic Effects in the Semisaturated Planar Tetracoordinated Carbon Molecule C7H4. Atoms 2022, 10, 10. [Google Scholar] [CrossRef]
- Yadav, K.; Lourderaj, U.; Priyakumar, U.D. Stereomutation in Tetracoordinate Centers via Stabilization of Planar Tetracoordinated Systems. Atoms 2021, 9, 79. [Google Scholar] [CrossRef]
- Stohrer, W.D.; Hoffmann, R. Bond-stretch isomerism and polytopal rearrangements in (CH)5+, (CH)5−, and (CH)4CO. J. Am. Chem. Soc. 1972, 94, 1661–1668. [Google Scholar] [CrossRef]
- Minkin, V.; Zefirov, N.; Korobov, M.; Averina, N.; Boganov, A.; Nivorozhkin, L. 1,3,5-Trimethyl Derivative of the Square Pyramidal Cation (Ch) 5+. Chem. Inf. Dienst. 1981, 12, 2616–2618. [Google Scholar]
- Krogh-Jespersen, K.; Chandrasekhar, J.; Schleyer, P.V.R. Geometries and relative energies of some C6H5+ and C5H5Si+ isomers. Pyramidal (nido) vs. planar, cyclic structures. J. Org. Chem. 1980, 45, 1608–1614. [Google Scholar] [CrossRef]
- Kenny, J.P.; Krueger, K.M.; Rienstra-Kiracofe, J.C.; Schaefer, H.F. C5H4: Pyramidane and its low-lying isomers. J. Phys. Chem. A 2001, 105, 7745–7750. [Google Scholar] [CrossRef]
- Lee, V.Y.; Gapurenko, O.A.; Ito, Y.; Meguro, T.; Sugasawa, H.; Sekiguchi, A.; Minyaev, R.M.; Minkin, V.I.; Herber, R.H.; Gornitzka, H. Pyramidanes: The Covalent Form of the Ionic Compounds. Organometallics 2016, 35, 346–356. [Google Scholar] [CrossRef]
- Gapurenko, O.A.; Minyaev, R.M.; Minkin, V.I. Silicon analogues of pyramidane: A quantum-chemical study. Mendeleev. Commun. 2012, 22, 8–10. [Google Scholar] [CrossRef]
- Moreno-Armenta, M.G.; Pearce, H.R.; Winter, P.; Cooksy, A.L. Computational search for metastable high-spin C5Hn (n = 4, 5, 6) species. Comput. Theor. Chem. 2018, 1140, 1–6. [Google Scholar] [CrossRef]
- Adewale, R.; da Silva, G. Kinetics of C5H4 isomer + H reactions and incorporation of C5Hx (x = 3–5) chemistry into a detailed chemical kinetic model. Combust. Flame 2021, 227, 227–237. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+ G basis set for first-row elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. A direct MP2 gradient method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Purvis, G.D. Many-body perturbation theory, coupled-pair many-electron theory, and the importance of quadruple excitations for the correlation problem. Int. J. Quantum. Chem. 1978, 14, 561–581. [Google Scholar] [CrossRef]
- Pople, J.; Krishnan, R.; Schlegel, H.; Binkley, J. Electron correlation theories and their application to the study of simple reaction potential surfaces. Int. J. Quantum. Chem. 1978, 14, 545–560. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Head-Gordon, M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Peterson, K.A.; Dunning, T.H., Jr. Accurate correlation consistent basis sets for molecular core–valence correlation effects: The second row atoms Al–Ar, and the first row atoms B–Ne revisited. J. Chem. Phys. 2002, 117, 10548–10560. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef]
- Frisch, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Matthews, D.A.; Cheng, L.; Harding, M.E.; Lipparini, F.; Stopkowicz, S.; Jagau, T.-C.; Szalay, P.G.; Gauss, J.; Stanton, J.F. Coupled-cluster techniques for computational chemistry: The CFOUR program package. J. Chem. Phys. 2020, 152, 214108. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software update: The ORCA program system, version 4.0. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, H.B.; Millam, J.M.; Iyengar, S.S.; Voth, G.A.; Daniels, A.D.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. J. Chem. Phys. 2001, 114, 9758–9763. [Google Scholar] [CrossRef]
- Walmsley, C.M.; Jewell, P.R.; Snyder, L.E.; Winnewisser, G. Detection of interstellar methyldiacetylene (CH3C4H) in dark dust cloud TMC1. Astron. Astrophys. 1984, 134, L11–L14. [Google Scholar]
- MacLeod, J.M.; Avery, L.W.; Broten, N.W. The detection of interstellar methyldiacetylene (CH3C4H). Astrophys. J. 1984, 282, L89–L92. [Google Scholar] [CrossRef]
- Araki, M.; Takano, S.; Sakai, N.; Yamamoto, S.; Oyama, T.; Kuze, N.; Tsukiyama, K. Long Carbon Chains in the Warm Carbon-chain-chemistry Source L1527: First Detection of C7H in Molecular Clouds. Astrophys. J. 2017, 847, 51. [Google Scholar] [CrossRef]
- Cabezas, C.; Fuentetaja, R.; Roueff, E.; Agúndez, M.; Tercero, B.; Marcelino, N.; Pardo, J.R.; de Vicente, P.; Cernicharo, J. New deuterated species in TMC-1: Detection of CH2DC4H with the QUIJOTE line survey. Astron. Astrophys. 2022, 657, L5. [Google Scholar] [CrossRef]
- Tsuneda, T.; Song, J.-W.; Suzuki, S.; Hirao, K. On Koopmans’ theorem in density functional theory. J. Chem. Phys. 2010, 133, 174101. [Google Scholar] [CrossRef] [PubMed]
- Merino, G.; Méndez-Rojas, M.A.; Beltrán, H.I.; Corminboeuf, C.; Heine, T.; Vela, A. Theoretical Analysis of the Smallest Carbon Cluster Containing a Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 2004, 126, 16160–16169. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Point-Group | B3LYP/6-311+G(d,p) | ωB97XD/6-311+G(d,p) | MP2/6-311+G(d,p) | fc-CCSD(T)/cc-pVTZ | NImag | Dipole Moment |
|---|---|---|---|---|---|---|---|
| Pent-1,3-diyne | C3v | 0 | 0 | 0 | 0 | 0 | 1.36 |
| py-1 | C4v | 55 | 42 | 43 | 44 | 0 | 1.83 |
| py-2 | Cs | 75 | 61 | 66 | 65 | 0 | 4.26 |
| ptC-1 | C2v | 143 | 140 | 146 | 152 | 4 | 0.57 |
| ptC-2 | C2v | 92 | 85 | 93 | 91 | 0 | 4.64 |
| ptC-3 | C2h | 142 | 139 | 150 | 146 | 1 | 0 |
| spiro-1 | D2d | 59 | 52 | 59 | 57 | 0 | 0 |
| spiro-2 | C2v | 108 | 102 | 109 | 105 | 2 | 3.40 |
| spiro-3 | C2 | 120 | 117 | 128 | 120 | 0 | 2.61 |
| spiro-4 | Cs | 97 | 90 | 102 | 97 | 0 | 3.57 |
| py-1 | py-2 | ptC-1 | ptC-2 | ptC-3 | spiro-1 | spiro-2 | spiro-3 | spiro-4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C1-C2 | 0.70 | C1-C2 | 1.07 | C1-C2 | 1.67 | C1-C2 | 1.56 | C1-C2 | 1.12 | C1-C2 | 1.88 | C1-C2 | 0.96 | C1-C2 | 1.06 | C1-C2 | 0.96 |
| C1-C3 | 0.70 | C1-C3 | 1.07 | C1-C3 | 0.98 | C1-C3 | 1.13 | C1-C3 | 0.72 | C1-C3 | 0.97 | C1-C3 | 1.01 | C1-C3 | 0.81 | C1-C3 | 1.14 |
| C1-C4 | 0.70 | C2-C3 | 0.56 | C2-C3 | 0.98 | C2-C3 | 1.13 | C2-C3 | 1.20 | C2-C3 | 0.97 | C2-C3 | 1.01 | C2-C3 | 1.13 | C2-C3 | 1.01 |
| C1-C5 | 0.70 | C3-C4 | 0.80 | C3-C4 | 0.98 | C3-C4 | 0.87 | C3-C4 | 1.20 | C3-C4 | 0.97 | C3-C4 | 0.97 | C3-C4 | 1.13 | C3-C4 | 0.87 |
| C2-C3 | 1.10 | C3-C5 | 0.80 | C3-C5 | 0.98 | C3-C5 | 0.87 | C3-C5 | 0.72 | C3-C5 | 0.97 | C3-C5 | 0.97 | C3-C5 | 1.06 | C3-C5 | 0.87 |
| C3-C4 | 1.10 | C4-C5 | 0.84 | C4-C5 | 1.67 | C4-C5 | 1.02 | C4-C5 | 1.12 | C4-C5 | 1.88 | C4-C5 | 2.44 | C4-C5 | 1.06 | C4-C5 | 1.99 |
| C4-C5 | 1.10 | C4-H8 | 1.03 | C1-H8 | 0.88 | C4-H8 | 0.91 | C1-H8 | 0.90 | C1-H9 | 0.90 | C1-H7 | 0.90 | C1-H8 | 0.90 | C2-H7 | 0.91 |
| C5-C2 | 1.10 | C4-H9 | 1.03 | C2-H9 | 0.88 | C4-H9 | 0.91 | C1-H9 | 0.90 | C2-H8 | 0.90 | C1-H8 | 0.90 | C1-H9 | 0.90 | C2-H9 | 0.91 |
| C2-H6 | 0.91 | C5-H6 | 0.91 | C4-H6 | 0.88 | C5-H6 | 0.91 | C5-H6 | 0.90 | C4-H6 | 0.90 | C2-H6 | 0.90 | C5-H6 | 0.90 | C4-H8 | 0.90 |
| C3-H7 | 0.91 | C5-H7 | 0.91 | C5-H7 | 0.88 | C5-H7 | 0.91 | C5-H7 | 0.90 | C5-H7 | 0.90 | C2-H9 | 0.90 | C5-H7 | 0.90 | C5-H6 | 0.90 |
| C4-H8 | 0.91 | C1-C2 | 0.90 | ||||||||||||||
| C5-H9 | 0.91 | C1-C3 | 0.89 | ||||||||||||||
| Molecules | BCP | Index | K(rc) | ρ(rc) | (∇2ρ(rc)) | G(rc) | V(rc) | H(rc) | ELF | G(rc)/V(rc) | G(rc)/ ρ(rc) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| py-1 | 8(H)--4(C) | 10 | 0.284 | 0.285 | −1.002 | 0.034 | −0.318 | −0.284 | 0.991 | −0.106 | 0.119 |
| 4(C)--1(C) | 11 | 0.106 | 0.169 | −0.021 | 0.100 | −0.206 | −0.106 | 0.685 | −0.487 | 0.595 | |
| 4(C)--3(C) | 12 | 0.257 | 0.274 | −0.631 | 0.099 | −0.356 | −0.257 | 0.918 | −0.278 | 0.361 | |
| py-2 | 9(H)--2(C) | 10 | 0.282 | 0.283 | −0.992 | 0.034 | −0.316 | −0.282 | 0.991 | −0.107 | 0.119 |
| 2(C)--4(C) | 11 | 0.220 | 0.253 | −0.507 | 0.094 | −0.314 | −0.220 | 0.905 | −0.298 | 0.371 | |
| 2(C)--1(C) | 12 | 0.233 | 0.256 | −0.560 | 0.093 | −0.326 | −0.233 | 0.910 | −0.285 | 0.364 | |
| ptC-1 | 7(H)--5(C) | 10 | 0.255 | 0.266 | −0.876 | 0.036 | −0.290 | −0.255 | 0.987 | −0.123 | 0.134 |
| 5(C)--3(C) | 12 | 0.177 | 0.225 | −0.245 | 0.116 | −0.292 | −0.177 | 0.811 | −0.395 | 0.513 | |
| 5(C)--4(C) | 14 | 0.364 | 0.327 | −0.899 | 0.140 | −0.504 | −0.364 | 0.910 | −0.277 | 0.427 | |
| ptC-2 | 6(H)--5(C) | 10 | 0.278 | 0.280 | −0.960 | 0.038 | −0.315 | −0.278 | 0.988 | −0.119 | 0.134 |
| 2(C)--1(C) | 12 | 0.434 | 0.358 | −1.118 | 0.154 | −0.588 | −0.434 | 0.919 | −0.262 | 0.430 | |
| 2(C)--3(C) | 13 | 0.161 | 0.231 | 0.091 | 0.183 | −0.344 | −0.161 | 0.649 | −0.533 | 0.795 | |
| 5(C)--3(C) | 14 | 0.189 | 0.230 | −0.375 | 0.096 | −0.285 | −0.189 | 0.872 | −0.335 | 0.414 | |
| 5(C)--4(C) | 16 | 0.218 | 0.251 | −0.513 | 0.089 | −0.307 | −0.218 | 0.911 | −0.291 | 0.356 | |
| ptC-3 | 6(H)--5(C) | 10 | 0.274 | 0.278 | −0.948 | 0.037 | −0.310 | −0.274 | 0.989 | −0.118 | 0.132 |
| 5(C)--4(C) | 12 | 0.278 | 0.284 | −0.695 | 0.104 | −0.381 | −0.278 | 0.920 | −0.272 | 0.365 | |
| 4(C)--3(C) | 13 | 0.237 | 0.250 | −0.426 | 0.131 | −0.368 | −0.237 | 0.827 | −0.355 | 0.522 | |
| spiro-1 | 8(H)--2(C) | 10 | 0.274 | 0.277 | −0.953 | 0.036 | −0.310 | −0.274 | 0.989 | −0.116 | 0.130 |
| 2(C)--3(C) | 11 | 0.203 | 0.243 | −0.378 | 0.109 | −0.312 | −0.203 | 0.862 | −0.348 | 0.447 | |
| 2(C)--1(C) | 12 | 0.403 | 0.341 | −0.909 | 0.176 | −0.578 | −0.403 | 0.881 | −0.304 | 0.515 | |
| spiro-2 | 9(H)--2(C) | 10 | 0.272 | 0.276 | −0.930 | 0.040 | −0.312 | −0.272 | 0.986 | −0.128 | 0.144 |
| 4(C)--5(C) | 12 | 0.599 | 0.423 | −1.476 | 0.230 | −0.829 | −0.599 | 0.898 | −0.277 | 0.544 | |
| 2(C)--3(C) | 13 | 0.243 | 0.266 | −0.583 | 0.098 | −0.341 | −0.243 | 0.913 | −0.286 | 0.367 | |
| 2(C)--1(C) | 15 | 0.171 | 0.222 | −0.334 | 0.087 | −0.258 | −0.171 | 0.878 | −0.338 | 0.392 | |
| spiro-3 | 2(C)--1(C) | 10 | 0.233 | 0.259 | −0.522 | 0.103 | −0.336 | −0.233 | 0.897 | −0.306 | 0.396 |
| 2(C)--3(C) | 11 | 0.277 | 0.274 | −0.578 | 0.132 | −0.409 | −0.277 | 0.862 | −0.323 | 0.483 | |
| 1(C)--8(H) | 12 | 0.270 | 0.276 | −0.934 | 0.037 | −0.307 | −0.270 | 0.988 | −0.120 | 0.134 | |
| 3(C)--1(C) | 14 | 0.160 | 0.216 | −0.157 | 0.121 | −0.282 | −0.160 | 0.772 | 0.430 | 0.561 | |
| spiro-4 | 8(H)--4(C) | 10 | 0.282 | 0.282 | −0.997 | 0.033 | −0.315 | −0.282 | 0.991 | −0.104 | 0.116 |
| 4(C)--5(C) | 11 | 0.454 | 0.362 | −1.017 | 0.200 | −0.654 | −0.454 | 0.874 | −0.306 | 0.553 | |
| 4(C)--3(C) | 13 | 0.170 | 0.221 | −0.236 | 0.111 | −0.281 | −0.170 | 0.814 | −0.395 | 0.502 | |
| 3(C)--2(C) | 15 | 0.219 | 0.252 | −0.463 | 0.103 | −0.322 | −0.219 | 0.886 | −0.320 | 0.411 | |
| 3(C)--1(C) | 16 | 0.294 | 0.295 | −0.735 | 0.111 | −0.405 | −0.294 | 0.920 | −0.273 | 0.375 |
| Molecules | NICS(1) |
|---|---|
| py-1 | −9.14 a; −10.43 b |
| py-2 | −4.72 a; −10.00 b |
| ptC-1 | 11.20 |
| ptC-2 | −18.87 |
| ptC-3 | −10.66 |
| spiro-1 | −5.38 |
| spiro-2 | 14.40 |
| spiro-3 | −11.82 |
| spiro-4 | −14.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Satpati, S.; Roy, T.; Giri, S.; Anoop, A.; Thimmakondu, V.S.; Ghosal, S. Structure and Bonding Patterns in C5H4 Isomers: Pyramidane, Planar Tetracoordinate Carbon, and Spiro Molecules. Atoms 2023, 11, 96. https://doi.org/10.3390/atoms11060096
Satpati S, Roy T, Giri S, Anoop A, Thimmakondu VS, Ghosal S. Structure and Bonding Patterns in C5H4 Isomers: Pyramidane, Planar Tetracoordinate Carbon, and Spiro Molecules. Atoms. 2023; 11(6):96. https://doi.org/10.3390/atoms11060096
Chicago/Turabian StyleSatpati, Sayon, Tarun Roy, Sandip Giri, Anakuthil Anoop, Venkatesan S. Thimmakondu, and Subhas Ghosal. 2023. "Structure and Bonding Patterns in C5H4 Isomers: Pyramidane, Planar Tetracoordinate Carbon, and Spiro Molecules" Atoms 11, no. 6: 96. https://doi.org/10.3390/atoms11060096
APA StyleSatpati, S., Roy, T., Giri, S., Anoop, A., Thimmakondu, V. S., & Ghosal, S. (2023). Structure and Bonding Patterns in C5H4 Isomers: Pyramidane, Planar Tetracoordinate Carbon, and Spiro Molecules. Atoms, 11(6), 96. https://doi.org/10.3390/atoms11060096