Metabolomics Analyses in High-Low Feed Efficient Dairy Cows Reveal Novel Biochemical Mechanisms and Predictive Biomarkers


Abstract
1. Introduction
2. Results
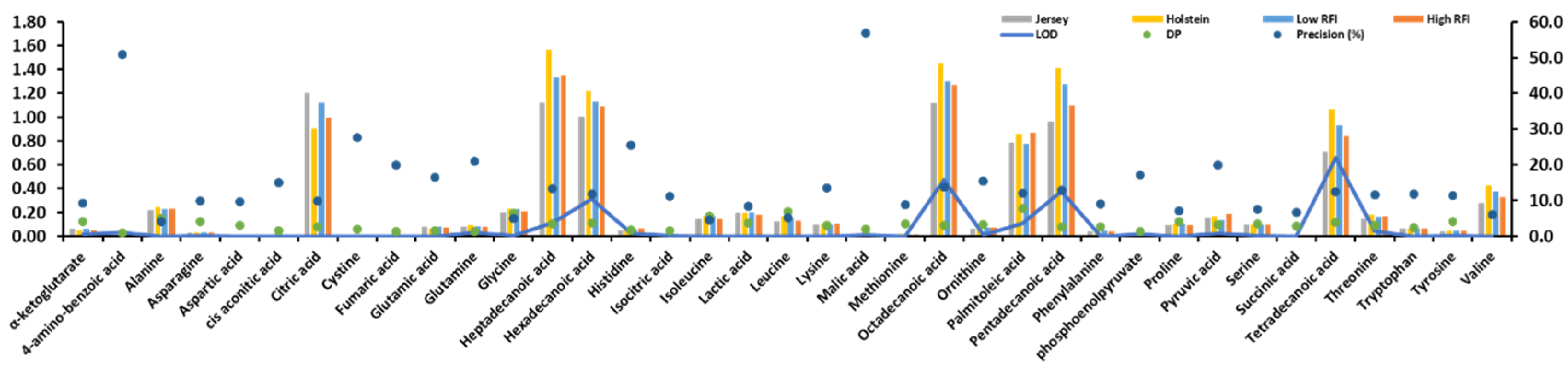
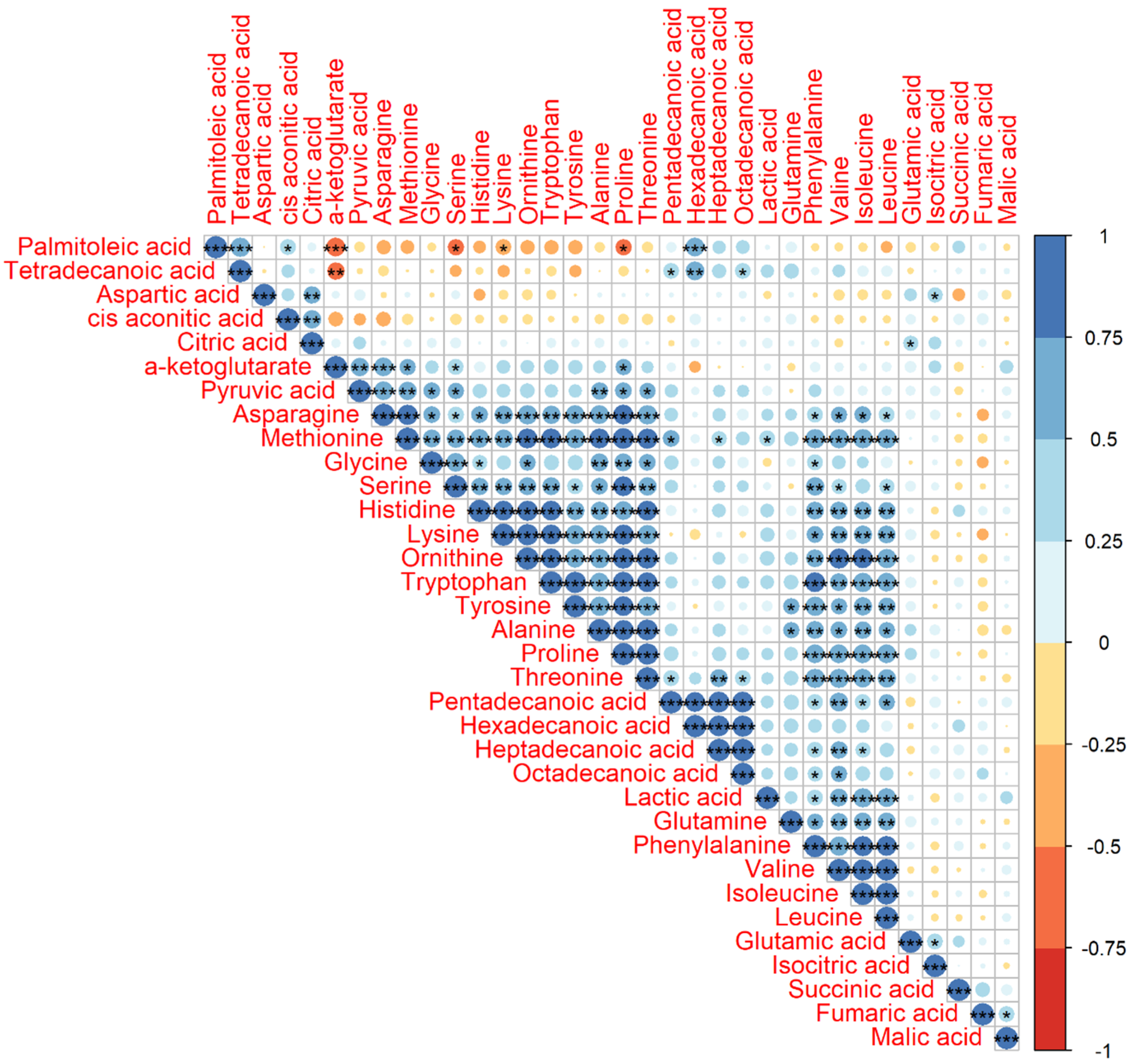
2.1. Statistics of the Identified Metabolites and their Pearson Correlation Coefficients
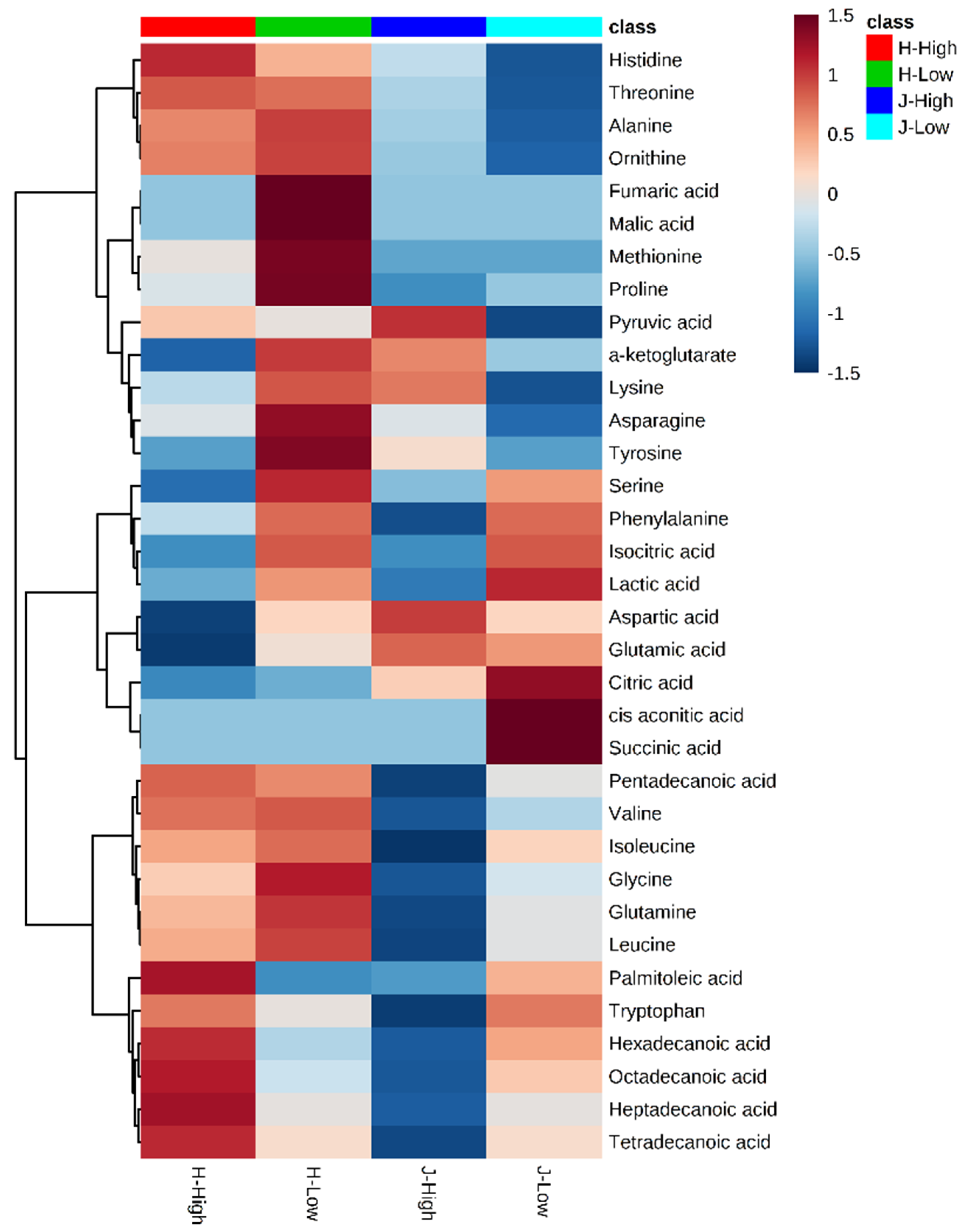
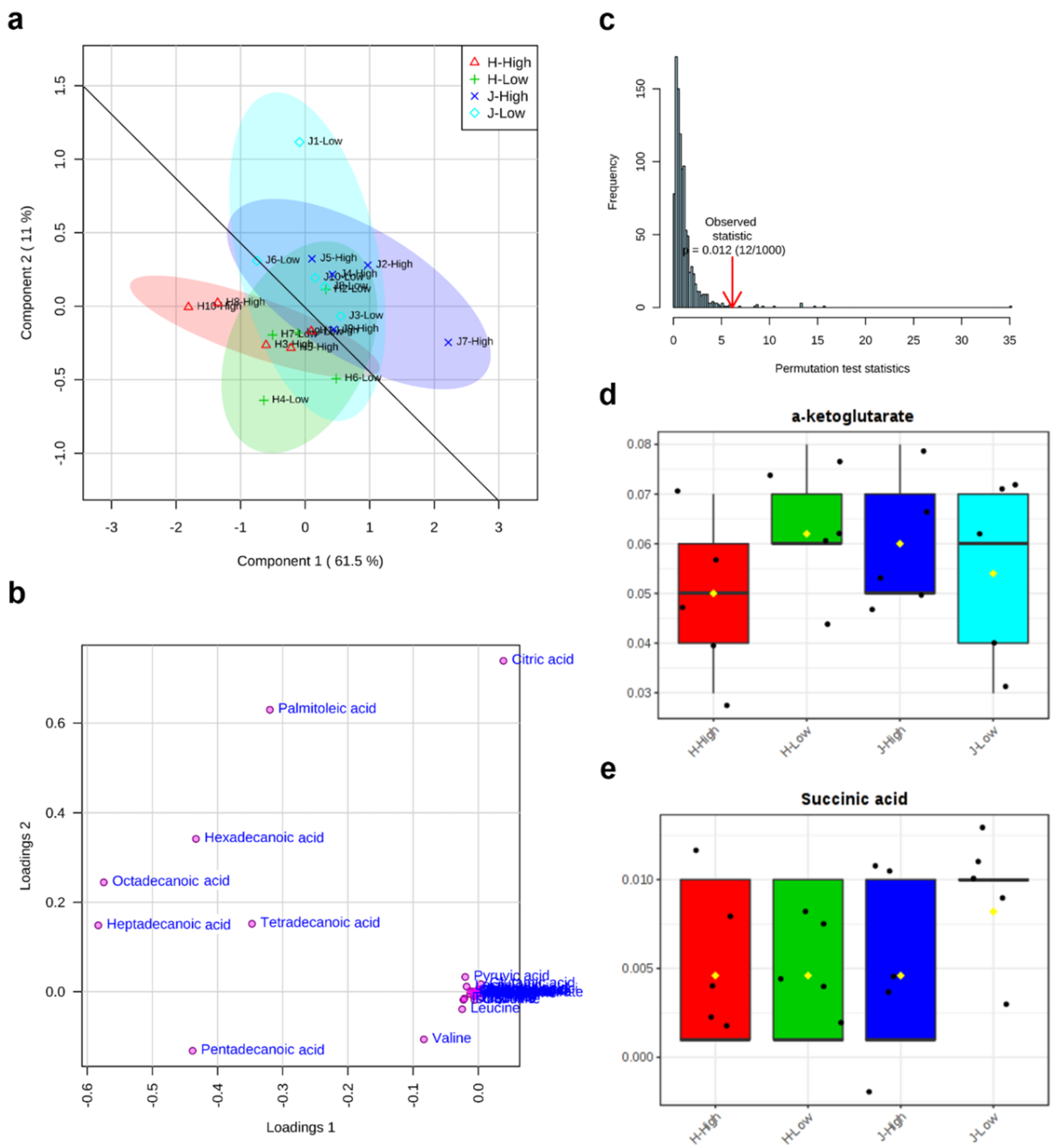
2.2. Metabolite Clusters and Comparisons between Low and High RFIs
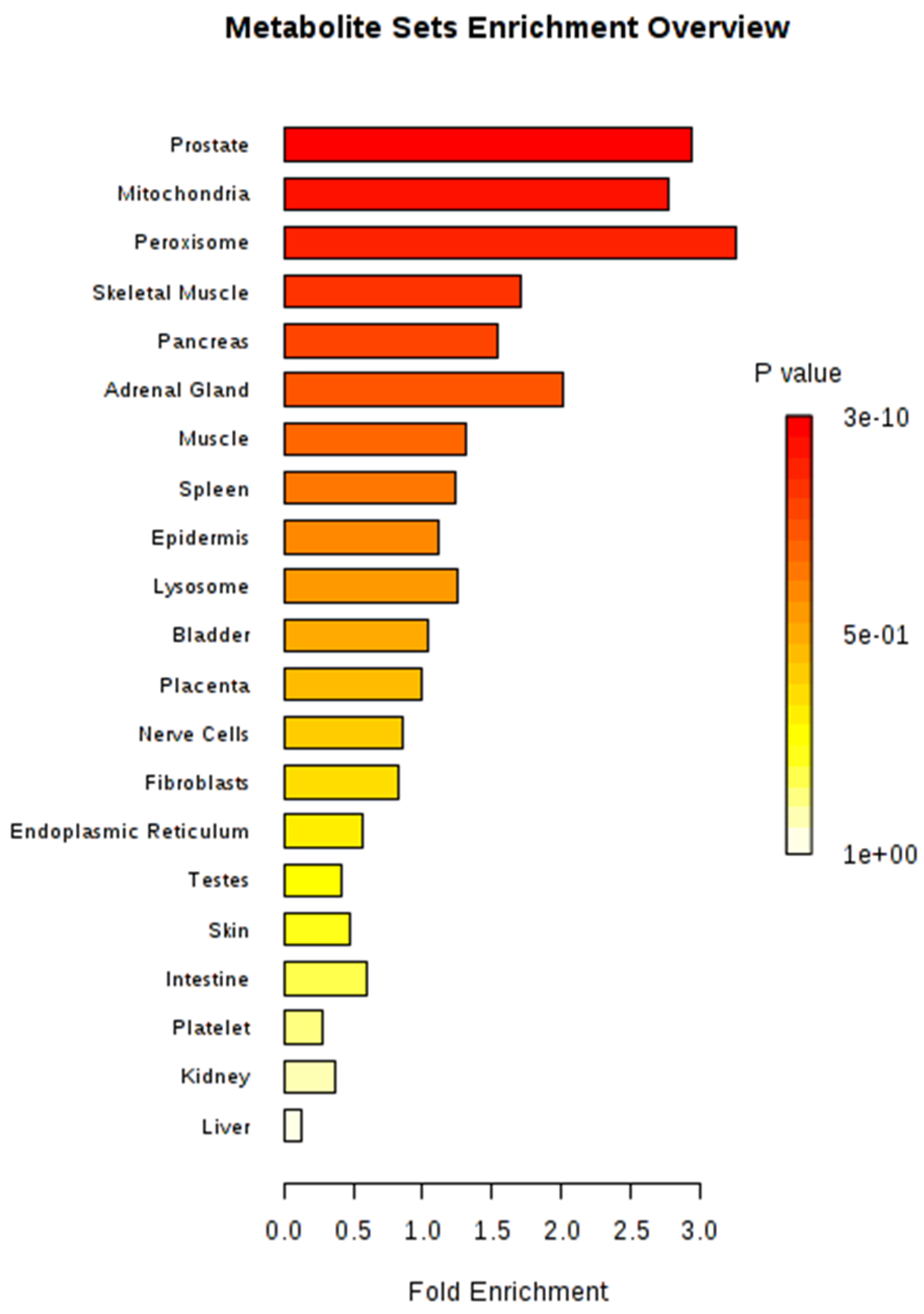
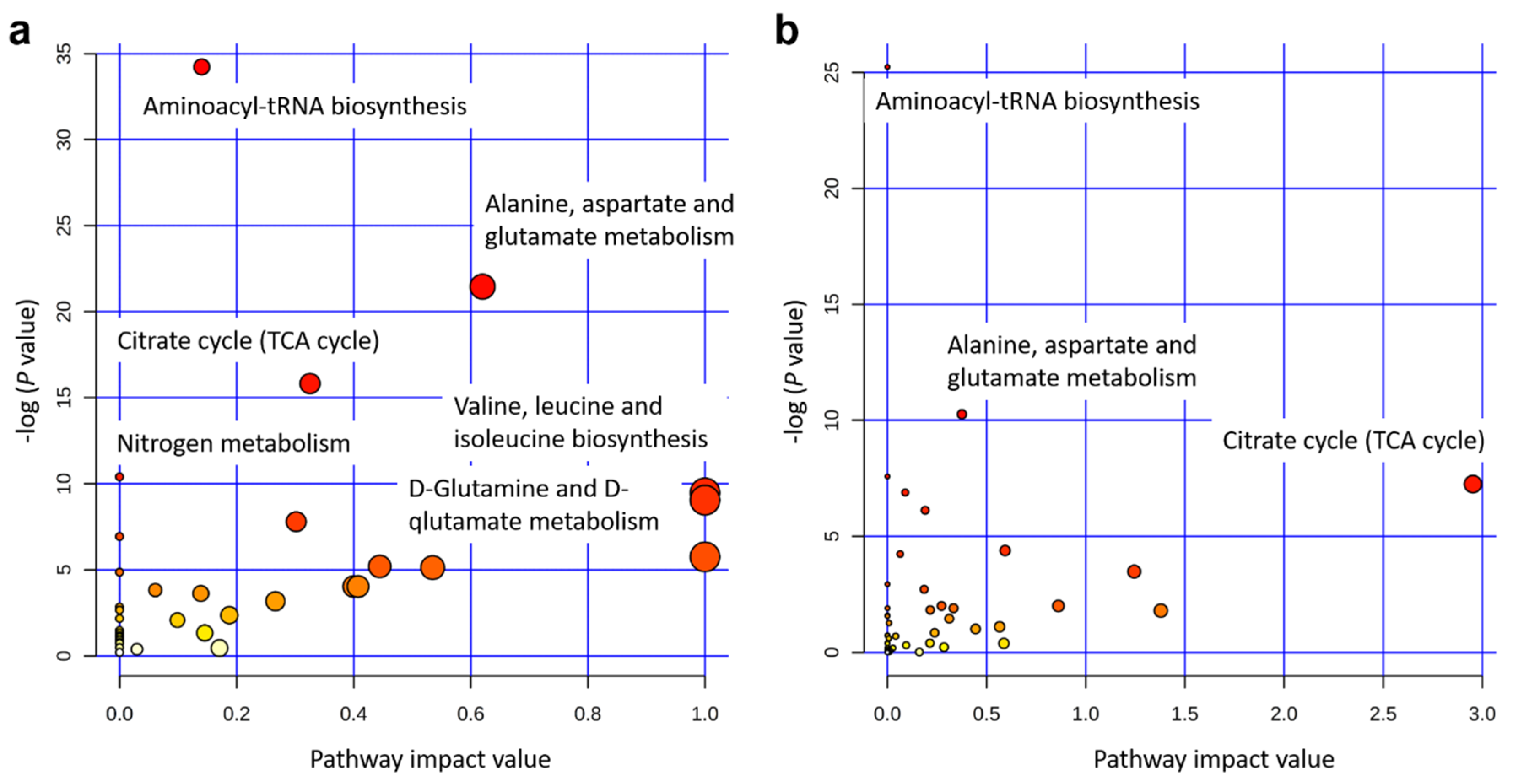
2.3. Significant Metabolic Enrichments, Pathways, and Networks
3. Discussion
3.1. Plasma Metabolites of Nordic Dairy Cattle
3.2. Key Metabolic Pathways after Single and Integrated Analysis
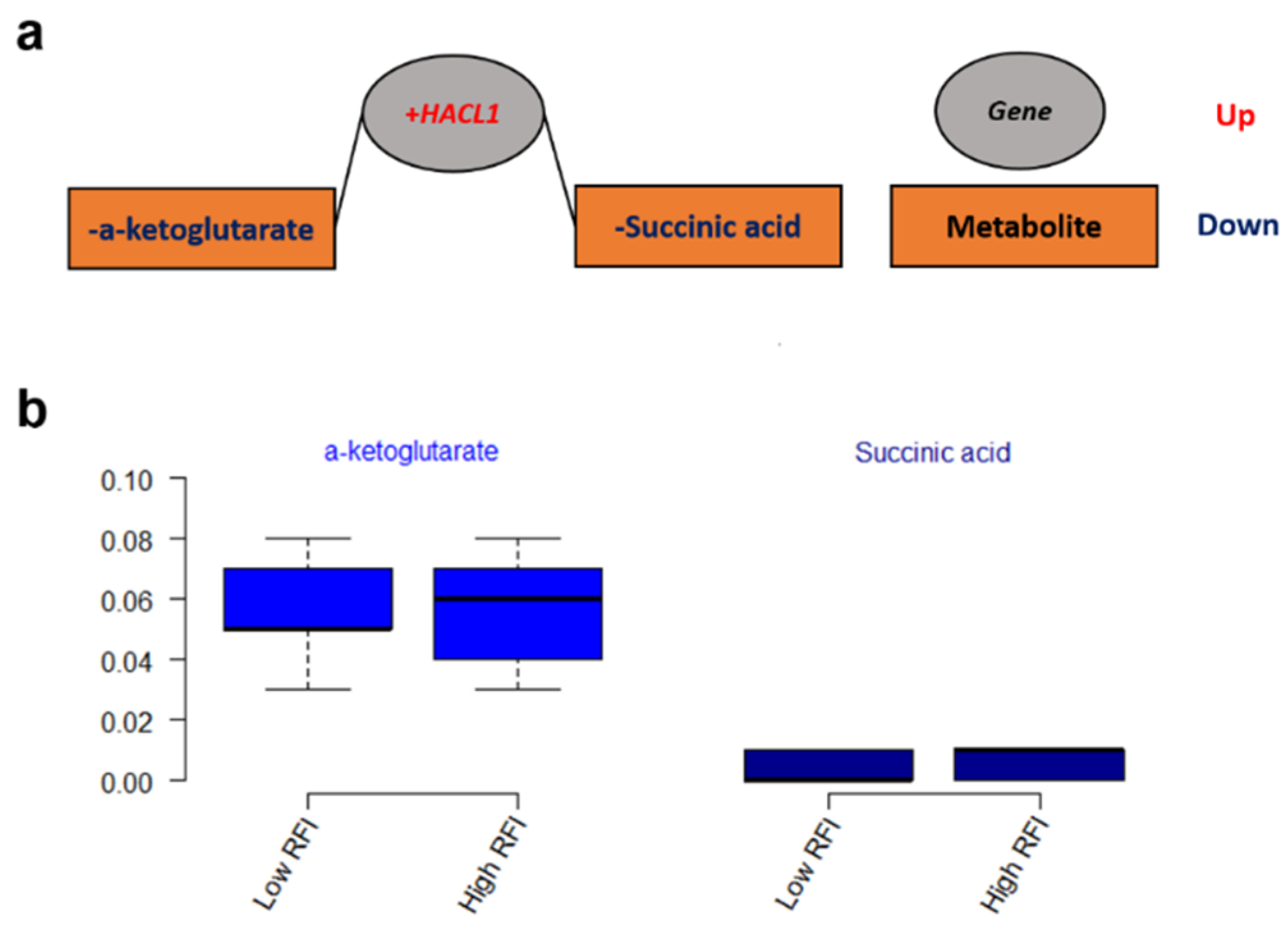
3.3. Metabolic Networks for Gene Expressions and Metabolites
3.4. Implications
4. Materials and Methods
4.1. Animals and Data
4.2. Metabolomics for Plasma
4.3. Statistical Analysis
4.4. Metabolite Enrichment and Pathway Characterization
4.5. Integration of Metabolomics and Transcriptomics Profiles in Low and High RFI Groups
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data and Material
Ethics Approval and Consent to Participate
Abbreviations
| ATP | Adenosine triphosphate |
| CD | Concentrated diets |
| Component 1 | First component |
| Component 2 | Second component |
| DCRC | Danish cattle research center |
| DMI | Dry matter intake |
| DP | Descriptive power |
| eQTL | Expression quantitative trait locus |
| FDR | False discovery rate |
| GABA | Gamma-aminobutyric acid |
| GC-MS | Gas chromatography - Mass spectrometry |
| GFE | Gross feed efficiency |
| GWAS | Genome-wide association study |
| HACL1 | 2-hydroxyacyl-CoA lyase 1 |
| ID | Identity card |
| LMDB | Livestock metabolome database |
| LOD | Limit of detection |
| logFC | Log of fold change |
| MCF | Methyl chloroformate |
| MSEA | Metabolite set enrichment analysis |
| MSI | Metabolomics Standards Initiative |
| NIST | National Institute of Standards and Technology |
| ORA | Over Representation Analysis |
| PARAFAC2 | PARAllel FACtor analysis 2 |
| PCC | Pearson correlation coefficient |
| PLS-DA | Partial least squares - discriminant analysis |
| QC | Quality control |
| RFI | Residual feed intake |
| SE | Standard error |
| SNP | Single nucleotide polymorphism |
| TCA cycle | Citrate cycle |
| TPP | Thiamin pyrophosphate |
| tRNA | Transfer RNA |
References
- Connor, E.E. Invited review: Improving feed efficiency in dairy production: Challenges and possibilities. Animal 2015, 9, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Connor, E.E.; Hutchison, J.L.; Olson, K.M.; Norman, H.D. Triennial lactation symposium: Opportunities for improving milk production efficiency in dairy cattle. J. Anim. Sci. 2012, 90, 1687–1694. [Google Scholar] [CrossRef] [PubMed]
- Koch, R.M.; Swiger, L.A.; Chambers, D.; Gregory, K.E. Efficiency of Feed Use in Beef Cattle. J. Anim. Sci. 1963, 22, 486–494. [Google Scholar] [CrossRef]
- Olijhoek, D.W.; Løvendahl, P.; Lassen, J.; Hellwing, A.L.F.; Höglund, J.K.; Weisbjerg, M.R.; Noel, S.; McLetan, F.; Højberg, O.; Lund, P. Methane production, rumen fermentation, and diet digestibility of Holstein and Jersey dairy cows being divergent in residual feed intake and fed at 2 forage-to-concentrate ratios. J. Dairy Sci. 2018, 101, 9926–9940. [Google Scholar] [CrossRef] [PubMed]
- Waghorn, G.C.; Woodward, S.L.; Tavendale, M.; Clark, D.A. Inconsistencies in rumen methane production—Effects of forage composition and animal genotype. Int. Congr. Ser. 2006, 1293, 115–118. [Google Scholar] [CrossRef]
- Kristensen, T.; Jensen, C.; Østergaard, S.; Weisbjerg, M.R.; Aaes, O.; Nielsen, N.I. Feeding, production, and efficiency of Holstein-Friesian, Jersey, and mixed-breed lactating dairy cows in commercial Danish herds. J. Dairy Sci. 2015, 98, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Grainger, C.; Goddard, M.E. A review of the effects of dairy breed on feed conversion efficiency—An opportunity lost? Proc. Bienn. Conf. Aust. Soc. Anim. Prod. 2004, 25, 77–80. [Google Scholar]
- Aikman, P.C.; Reynolds, C.K.; Beever, D.E. Diet Digestibility, Rate of Passage, and Eating and Rumination Behavior of Jersey and Holstein Cows. J. Dairy Sci. 2008, 91, 1103–1114. [Google Scholar] [CrossRef]
- Shetty, N.; Løvendahl, P.; Lund, M.S.; Buitenhuis, A.J. Prediction and validation of residual feed intake and dry matter intake in Danish lactating dairy cows using mid-infrared spectroscopy of milk. J. Dairy Sci. 2016, 100, 253–264. [Google Scholar] [CrossRef]
- Li, B.; Berglund, B.; Fikse, W.F.; Lassen, J.; Lidauer, M.H.; Mäntysaari, P.; Løvendahl, P. Neglect of lactation stage leads to naive assessment of residual feed intake in dairy cattle. J. Dairy Sci. 2017, 100, 9076–9084. [Google Scholar] [CrossRef]
- Salleh, S.M.; Mazzoni, G.; Løvendahl, P.; Kadarmideen, H.N. Gene co-expression networks from RNA sequencing of dairy cattle identifies genes and pathways affecting feed efficiency. BMC Bioinform. 2018, 19, 513. [Google Scholar] [CrossRef] [PubMed]
- Salleh, S.M.; Mazzoni, G.; Nielsen, M.O.; Løvendahl, P.; Kadarmideen, H.N. Identification of Expression QTLs Targeting Candidate Genes for Residual Feed Intake in Dairy Cattle Using Systems Genomics. J. Genet. Genome Res. 2018, 5, 035. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. “Metabonomics”: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Kenéz, Á.; Dänicke, S.; Rolle-Kampczyk, U.; von Bergen, M.; Huber, K. A metabolomics approach to characterize phenotypes of metabolic transition from late pregnancy to early lactation in dairy cows. Metabolomics 2016, 12, 165. [Google Scholar] [CrossRef]
- Guo, Y.S.; Tao, J.Z. Metabolomics and pathway analyses to characterize metabolic alterations in pregnant dairy cows on D 17 and D 45 after AI. Sci. Rep. 2018, 8, 5973. [Google Scholar] [CrossRef]
- Mazzoni, G.; Lund, P.; Løvendahl, P.; Salleh, M.S.; Höglund, J.K.; Olijhoek, D.W.; Kadarmideen, H.N. RNA-Seq transcriptomics and pathway analyses reveal potential regulatory genes and molecular mechanisms in high- and low-residual feed intake in Nordic dairy cattle. BMC Genom. 2017, 18, 258. [Google Scholar]
- Pryce, J.; Arias, J.; Bowman, P.; Davis, S.; Macdonald, K.; Waghorn, G.; Wales, W.; Williams, Y.; Spelman, R.; Hayes, B.; et al. Accuracy of genomic predictions of residual feed intake and 250-day body weight in growing heifers using 625,000 single nucleotide polymorphism markers. J. Dairy Sci. 2012, 95, 2108–2119. [Google Scholar] [CrossRef]
- Connor, E.E.; Hutchison, J.L.; Norman, H.D.; Olson, K.M.; Van Tassell, C.; Leith, J.M.; Baldwin, R.L. Use of residual feed intake in Holsteins during early lactation shows potential to improve feed efficiency through genetic selection. J. Anim. Sci. 2013, 91, 3978–3988. [Google Scholar] [CrossRef]
- Gohlke, R.S. Time-of-Flight Mass Spectrometry and Gas-Liquid Partition Chromatography. Anal. Chem. 1959, 31, 535–541. [Google Scholar] [CrossRef]
- Sparkman, O.D.; Penton, Z.; Kitson, F.G. Gas Chromatography and Mass Spectrometry: A Practical Guide; Elsevier Science, Elsevier Ltd.: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Zhou, Z.; Loor, J.J.; Piccioli-Cappelli, F.; Librandi, F.; Lobley, G.E.; Trevisi, E. Circulating amino acids in blood plasma during the peripartal period in dairy cows with different liver functionality index. J. Dairy Sci. 2016, 99, 2257–2267. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Viant, M.R.; Kurland, I.J.; Jones, M.R.; Dunn, W.B. How close are we to complete annotation of metabolomes? Curr. Opin. Chem. Biol. 2017, 36, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Lismont, C.; Walton, P. The peroxisome-mitochondria connection: How and why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef] [PubMed]
- Demarquoy, J.; Le Borgne, F.; Demarquoy, F.J.; Le Borgne, O. Crosstalk between mitochondria and peroxisomes. World J. Biol. Chem. 2015, 26, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Ali Goldansaz, S.; Chi Guo, A.; Sajed, T.; Steele, M.A.; Plastow, G.S.; Wishart, D.S. Livestock metabolomics and the livestock metabolome: A systematic review. PLoS ONE 2017, 12, 0177675. [Google Scholar] [CrossRef] [PubMed]
- Karisa, B.; Thomson, J.; Wang, Z.; Li, C.; Montanholi, Y.; Miller, S.; Moore, S.; Plastow, G.; Plastow, G. Plasma metabolites associated with residual feed intake and other productivity performance traits in beef cattle. Livest. Sci. 2014, 165, 200–211. [Google Scholar] [CrossRef]
- Mayes, P.; Bender, D. The citric acid cycle: The catabolism of AcetylCoA. In Harper’s Illustrated Biochemistry; Murray, R.K., Granner, D.K., Mayes, P.A., Rodwell, V., Eds.; Lange Medical Books; Mc Graw Hill Companies: New York, NY, USA, 2003; pp. 130–135. [Google Scholar]
- Kyriacou, S.V.; Deutscher, M.P. An Important Role for the Multienzyme Aminoacyl-tRNA Synthetase Complex in Mammalian Translation and Cell Growth. Mol. Cell 2008, 29, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Bergert, M.; Walther, A.; Suter, B. Double-sieving-defective aminoacyl-tRNA synthetase causes protein mistranslation and affects cellular physiology and development. Nat. Commun. 2014, 27, 5650. [Google Scholar] [CrossRef] [PubMed]
- Antonellis, A.; Ellsworth, R.E.; Sambuughin, N.; Puls, I.; Abel, A.; Lee-Lin, S.-Q.; Jordanova, A.; Kremensky, I.; Christodoulou, K.; Middleton, L.T.; et al. Glycyl tRNA Synthetase Mutations in Charcot-Marie-Tooth Disease Type 2D and Distal Spinal Muscular Atrophy Type, V. Am. J. Hum. Genet. 2003, 72, 1293–1295. [Google Scholar] [CrossRef]
- Jordanova, A.; Irobi, J.; Thomas, F.P.; Van Dijck, P.; Meerschaert, K.; Dewil, M.; Dierick, I.; Jacobs, A.; De Vriendt, E.; Guergueltcheva, V.; et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet. 2006, 38, 197–202. [Google Scholar] [CrossRef]
- Wu, N.; Yang, M.; Gaur, U.; Xu, H.; Yao, Y.; Li, D. Alpha-ketoglutarate: Physiological functions and applications. Biomol. Ther. 2016, 24, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim. Biophys. Acta-Bioenerg. 2016, 1857, 1086–1101. [Google Scholar] [CrossRef] [PubMed]
- Soghomonian, J.J.; Martin, D.L. Two isoforms of glutamate decarboxylase: Why? Trends Pharmacol. Sci. 1998, 19, 500–505. [Google Scholar] [CrossRef]
- Do, D.N.; Ostersen, T.; Strathe, A.B.; Mark, T.; Jensen, J.; Kadarmideen, H.N. Genome-wide association and systems genetic analyses of residual feed intake, daily feed consumption, backfat and weight gain in pigs. BMC Genet. 2014, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Kadarmideen, H.N. Genomics to Systems Biology in Animal and Veterinary Sciences: Progress, Lessons and Opportunities. Livest. Sci. 2014, 166, 232–248. [Google Scholar] [CrossRef]
- Suravajhala, P.; Kogelman, L.J.A.; Kadarmideen, H.N. Multi-omic data integration and analysis using systems genomics approaches: Methods and applications in animal production, health and welfare. Genet. Sel. Evol. 2016, 48, 38. [Google Scholar] [CrossRef]
- Xia, J.; Broadhurst, D.I.; Wilson, M.; Wishart, D.S. Translational biomarker discovery in clinical metabolomics: An introductory tutorial. Metabolomics 2013, 9, 280–299. [Google Scholar] [CrossRef] [PubMed]
- Tempelman, R.; Spurlock, D.; Coffey, M.; Veerkamp, R.; Armentano, L.; Weigel, K.; De Haas, Y.; Staples, C.; Connor, E.; Lu, Y.; et al. Heterogeneity in genetic and nongenetic variation and energy sink relationships for residual feed intake across research stations and countries. J. Dairy Sci. 2015, 98, 2013–2026. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef]
- Kiers, H.A.L.; ten Berge, J.M.F.; Bro, R. PARAFAC2—Part I. A direct fitting algorithm for the PARAFAC2 model. J. Chemom. Soc. 1999, 13, 275–294. [Google Scholar] [CrossRef]
- Ward, J.H. Hierarchical Grouping to Optimize an Objective Function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. MetPA: A web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010, 26, 2342–2344. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat. Protoc. 2011, 6, 743–760. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RFI | Amino Acid (Mean ± SE) | Tricarboxylic Acid (Mean ± SE) | Fatty Acid (Mean ± SE) | All 26 Metabolite (Mean ± SE) |
|---|---|---|---|---|
| Low | 0.12 ± 0.03 | 0.29 ± 0.12 | 1.10 ± 0.12*** | 0.38 ± 0.15*** |
| High | 0.11 ± 0.03 | 0.31 ± 0.14 | 1.52 ± 0.23*** | 0.48 ± 0.22*** |
| Metabolite (Mean ± SE) | Leucine | Ornithine | Pentadecanoic Acid | Valine |
|---|---|---|---|---|
| Breed | −0.05 ± 0.02* | −0.02 ± 0.01* | −0.51 ± 0.15** | 0.33 ± 0.05*** |
| RFI | 0.02 ± 0.01 (P = 0.06) | −0.001 ± 0.01 (P = 0.7) | 0.16 ± 0.08 (P = 0.07) | 0.04 ± 0.02 (P = 0.09) |
| Pathway Name | Match Status | P Value | -log (P Value) | FDR | Impact |
|---|---|---|---|---|---|
| Aminoacyl-tRNA biosynthesis | 17/64 | 1.4 × 10−15 | 34 | 1.1 × 10−13 | 0.14 |
| Alanine, aspartate, and glutamate metabolism | 9/23 | 4.8 × 10−10 | 21 | 2.0 × 10−8 | 0.62 |
| Citrate cycle (TCA cycle) | 7/20 | 1.4 × 10−7 | 16 | 3.7 × 10−6 | 0.33 |
| Nitrogen metabolism | 4/9 | 3.1 × 10−5 | 10 | 6.2 × 10−4 | 0 |
| Valine, leucine, and Isoleucine biosynthesis | 4/11 | 7.7 × 10−5 | 9.5 | 0.0013 | 1.0 |
| D-Glutamine and D-glutamate metabolism | 3/5 | 1.2 × 10−4 | 9.1 | 0.0016 | 1.0 |
| Arginine and proline metabolism | 6/44 | 4.1 × 10−4 | 7.8 | 0.0048 | 0.30 |
| Butanoate metabolism | 4/20 | 9.8 × 10−4 | 6.9 | 0.0099 | 0 |
| Phenylalanine, tyrosine, and tryptophan biosynthesis | 2/4 | 0.0032 | 5.8 | 0.029 | 1.0 |
| Glyoxylate and dicarboxylate metabolism | 3/16 | 0.0055 | 5.2 | 0.044 | 0.44 |
| Glycine, serine, and threonine metabolism | 4/32 | 0.0059 | 5.1 | 0.044 | 0.53 |
| Cyanoamino acid metabolism | 2/6 | 0.0077 | 4.9 | 0.052 | 0 |
| Methane metabolism | 2/9 | 0.018 | 4.0 | 0.10 | 0.4 |
| Phenylalanine metabolism | 2/9 | 0.018 | 4.0 | 0.10 | 0.41 |
| Glutathione metabolism | 3/26 | 0.021 | 3.8 | 0.12 | 0.061 |
| Cysteine and methionine metabolism | 3/28 | 0.027 | 3.6 | 0.13 | 0.14 |
| Histidine metabolism | 2/14 | 0.042 | 3.2 | 0.20 | 0.27 |
| Breed | RFI | Actual RFI Value | Cow ID | Parity | Breed | RFI | Actual RFI Value | Cow ID | Parity |
|---|---|---|---|---|---|---|---|---|---|
| Jersey | Low | 0.80 | J1-Low | 1 | Holstein | Low | −0.03 | H2-Low | 1 |
| 2.23 | J3-Low | 3 | 0.10 | H4-Low | 2 | ||||
| 0.94 | J6-Low | 3 | 0.70 | H6-Low | 3 | ||||
| 0.46 | J8-Low | 2 | 0.89 | H7-Low | 2 | ||||
| 0.49 | J10-Low | 1 | 0.41 | H9-Low | 1 | ||||
| High | −0.40 | J2-High | 1 | High | −1.10 | H1-High | 1 | ||
| −0.04 | J4-High | 3 | 0.05 | H3-High | 3 | ||||
| −1.05 | J5-High | 3 | −0.62 | H5-High | 3 | ||||
| −1.71 | J7-High | 2 | −1.05 | H8-High | 2 | ||||
| −0.51 | J9-High | 1 | −0.40 | H10-High | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Kadarmideen, H.N. Metabolomics Analyses in High-Low Feed Efficient Dairy Cows Reveal Novel Biochemical Mechanisms and Predictive Biomarkers. Metabolites 2019, 9, 151. https://doi.org/10.3390/metabo9070151
Wang X, Kadarmideen HN. Metabolomics Analyses in High-Low Feed Efficient Dairy Cows Reveal Novel Biochemical Mechanisms and Predictive Biomarkers. Metabolites. 2019; 9(7):151. https://doi.org/10.3390/metabo9070151
Chicago/Turabian StyleWang, Xiao, and Haja N. Kadarmideen. 2019. "Metabolomics Analyses in High-Low Feed Efficient Dairy Cows Reveal Novel Biochemical Mechanisms and Predictive Biomarkers" Metabolites 9, no. 7: 151. https://doi.org/10.3390/metabo9070151
APA StyleWang, X., & Kadarmideen, H. N. (2019). Metabolomics Analyses in High-Low Feed Efficient Dairy Cows Reveal Novel Biochemical Mechanisms and Predictive Biomarkers. Metabolites, 9(7), 151. https://doi.org/10.3390/metabo9070151