Computational Methods for the Discovery of Metabolic Markers of Complex Traits


Abstract
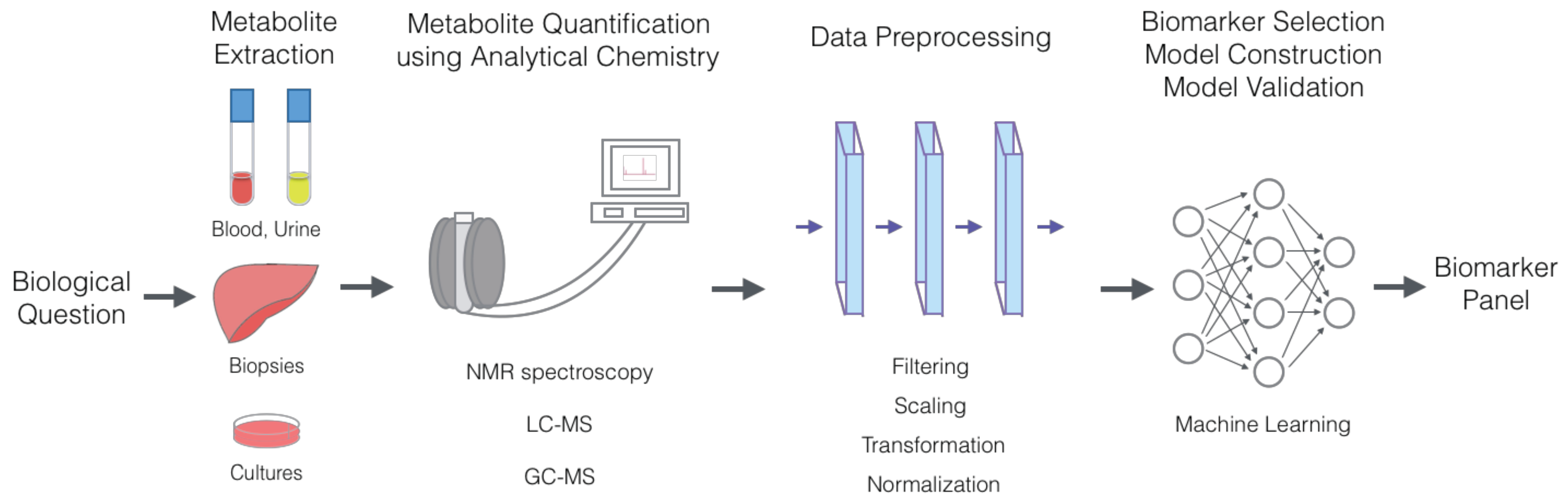
1. Introduction
2. Metabolic Marker Discovery
3. Current Research in Metabolomics, Complex Diseases and Computational Methods
3.1. Alzheimer’s Disease
3.2. Breast Cancer
3.3. Osteoarthritis
4. Advanced Learning Methods for Metabolic Marker Discovery
4.1. Ensemble Learning
4.2. Artificial Neural Networks
4.3. Genetic Programming
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NMR | nuclear magnetic resonance |
| MS | mass spectrometry |
| LC-MS | liquid chromatography-mass spectrometry |
| PLS-DA | partial least squares-discriminant analysis |
| K-OPLS | kernel-based orthogonal projections to latent structures |
| PCA | principal component analysis |
| SVM | support vector machine |
| RF | random forest |
| GBM | gradient boosting machine |
| ANN | artificial neural networks |
| AUC | area under the curve |
| ANOVA | analysis of variance |
| Aβ | amyloid beta |
| AD | Alzheimer’s disease |
| MCI | mild cognitive impairment |
| ADNI | Alzheimer’s Disease Neuroimaging Initiative |
| BLSA | Baltimore Longitudinal Study of Aging |
| CSF | cerebrospinal fluid |
| GA | genetic algorithm |
| ES | evolution strategy |
| EA | evolutionary algorithm |
| GP | genetic programming |
| ATP | adenosine 5′-triphosphate |
References
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Kaddurah-Daouk, R.; Kristal, B.S.; Weinshilboum, R.M. Metabolomics: A Global Biochemical Approach to Drug Response and Disease. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 653–683. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef] [PubMed]
- Craig, J. Complex diseases: Research and applications. Nat. Educ. 2008, 1, 184. [Google Scholar]
- Varma, V.R.; Oommen, A.M.; Varma, S.; Casanova, R.; An, Y.; Andrews, R.M.; O’Brien, R.; Pletnikova, O.; Troncoso, J.C.; Toledo, J.; et al. Brain and blood metabolite signatures of pathology and progression in Alzheimer disease: A targeted metabolomics study. PLoS Med. 2018, 15, e1002482. [Google Scholar] [CrossRef] [PubMed]
- Alakwaa, F.M.; Chaudhary, K.; Garmire, L.X. Deep Learning Accurately Predicts Estrogen Receptor Status in Breast Cancer Metabolomics Data. J. Proteome Res. 2018, 17, 337–347. [Google Scholar] [CrossRef]
- Hu, T.; Oksanen, K.; Zhang, W.; Randell, E.; Furey, A.; Sun, G.; Zhai, G. An evolutioanry learning and network approach to identifying key metabolites for osteoarthritis. PLoS Comput. Biol. 2018, 14, e1005986. [Google Scholar] [CrossRef]
- Alonso, A.; Marsal, S.; Julià, A. Analytical methods in untargeted metabolomics: State of the art in 2015. Front. Bioeng. Biotechnol. 2015, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Broadhurst, D.I.; Wilson, M.; Wishart, D.S. Translational biomarker discovery in clinical metabolomics: An introductory tutorial. Metabolomics 2013, 9, 280–299. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O.; Robertson, D.; Griffin, J.; van der Werf, M.; Nikolau, B.; Morrison, N.; Sumner, L.W.; Goodacre, R.; Hardy, N.W.; Taylor, C.; et al. The metabolomics standards initiative (MSI). Metabolomics 2007, 3, 175–178. [Google Scholar] [CrossRef]
- Spicer, R.A.; Salek, R.; Steinbeck, C. A decade after the metabolomics standards initiative it’s time for a revision. Sci. Data 2017, 4, 170138. [Google Scholar] [CrossRef] [PubMed]
- Broadhurst, D.I.; Kell, D.B. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2006, 2, 171–196. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.Z.; Begley, P.; Mullard, G.; Hollywood, K.A.; Bishop, P.N. Introduction to metabolomics and its applications in ophthalmology. Eye (Lond. Engl.) 2016, 30, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Rockel, J.S.; Zhang, W.; Shestopaloff, K.; Likhodii, S.; Sun, G.; Furey, A.; Randell, E.; Sundararajan, K.; Gandhi, R.; Zhai, G.; et al. A classification modelling approach for determining metabolite signatures in osteoarthritis. PLoS ONE 2018, 13, e0199618. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Semiz, S.; van der Lee, S.J.; van der Spek, A.; Verhoeven, A.; van Klinken, J.B.; Sijbrands, E.; Harms, A.C.; Hankemeier, T.; van Dijk, K.W.; et al. Metabolomics based markers predict type 2 diabetes in a 14-year follow-up study. Metab. Off. J. Metab. Soc. 2017, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic Quotient Normalization as Robust Method to Account for Dilution of Complex Biological Mixtures. Application in 1H NMR Metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Åstrand, M.; Speed, T.P. A Comparison of Normalization Methods for High Density Oligonucleotide Array Data Based on Variance and Bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Hoefer, I.E.; Steffens, S.; Ala-Korpela, M.; Back, M.; Badimon, L.; Bochaton-Piallat, M.L.; Boulanger, C.M.; Caligiuri, G.; Dimmeler, S.; Egido, J.; et al. Novel methodologies for biomarker discovery in atherosclerosis. Eur. Heart J. 2015, 36, 2635–2642. [Google Scholar] [CrossRef] [PubMed]
- Kaddurah-Daouk, R.; Weinshilboum, R. Metabolomic signatures for drug response phenotypes: Pharmacometabolomics enables precision medicine. Clin. Pharmacol. Ther. 2015, 98, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Sun, H.; Yan, G.; Wang, P.; Wang, X. Mass spectrometry-based metabolomics: Applications to biomarker and metabolic pathway research. Biomed. Chromatogr. 2016, 30, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Guyon, I.; Elisseeff, A. An introduction to variable and feature selection. J. Mach. Learn. Res. 2003, 3, 1157–1182. [Google Scholar]
- Saeys, Y.; Inza, I.; Larranaga, P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef] [PubMed]
- Cavill, R.; Keun, H.C.; Holmes, E.; Lindon, J.C.; Nicholson, J.K.; Ebbels, T.M.D. Genetic algorithms for simultaneous variable and sample selection in metabolomics. Bioinformatics 2009, 25, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Grissa, D.; Petera, M.; Brandolini, M.; Napoli, A.; Comte, B.; Pujos-Guillot, E. Feature selection methods for early predictive biomarker discovery using untargeted metabolomic data. Front. Mol. Biosci. 2016, 8, 30. [Google Scholar] [CrossRef]
- Ribeiro, M.T.; Singh, S.; Guestrin, C. “Why should I trust you?”: Explaining the predictions of any classifier. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 1135–1144. [Google Scholar]
- Yu, M.K.; Ma, J.; Fisher, J.; Kreisberg, J.F.; Raphael, B.J.; Ideker, T. Visible machine learning for biomedicine. Cell 2018, 173, 1562–1565. [Google Scholar] [CrossRef]
- Choromanska, A.; Henaff, M.; Mathieu, M.; Ben Arous, G.; LeCun, Y. The Loss Surfaces of Multilayer Networks. arXiv, 2014; arXiv:1412.0233. [Google Scholar]
- Bird, T.D. Alzheimer Disease Overview. GeneReviews®. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1161 (accessed on 4 April 2019).
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Cummings, J. Lessons Learned from Alzheimer Disease: Clinical Trials with Negative Outcomes. Clin. Transl. Sci. 2018, 11, 147–152. [Google Scholar] [CrossRef]
- Han, X.; Holtzman, D.M.; McKeel, D.W., Jr.; Kelley, J.; Morris, J.C. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: Potential role in disease pathogenesis. J. Neurochem. 2002, 82, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Rozen, S.; Boyle, S.H.; Hellegers, C.; Cheng, H.; Burke, J.R.; Welsh-Bohmer, K.A.; Doraiswamy, P.M.; Kaddurah-Daouk, R. Metabolomics in early Alzheimer’s disease: Identification of altered plasma sphingolipidome using shotgun lipidomics. PLoS ONE 2011, 6, e21643. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhou, Y.; Huang, F.J.; Tang, H.D.; Xu, X.H.; Liu, J.J.; Wang, Y.; Deng, Y.L.; Ren, R.J.; Xu, W.; et al. Plasma Metabolite Profiles of Alzheimer’s Disease and Mild Cognitive Impairment. J. Proteome Res. 2014, 13, 2649–2658. [Google Scholar] [CrossRef]
- Sabbagh, M.N.; Lue, L.F.; Fayard, D.; Shi, J. Increasing Precision of Clinical Diagnosis of Alzheimer’s Disease Using a Combined Algorithm Incorporating Clinical and Novel Biomarker Data. Neurol. Ther. 2017, 6, 83–95. [Google Scholar] [CrossRef]
- Siegel, R.; Miller, K.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Schifmann, J.; Fisher, P.; Gibbs, P. Early detection of cancer: Past, present, and future. Am. Soc. Clin. Oncol. Educ. Book 2015, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Catalona, W.; Richie, J.; Ahmann, F.; Hudson, M.; Scardino, P.; Flanigan, R.; DeKernion, J.; Ratliff, T.; Kavoussi, L.; Dalkin, B.; et al. Comparison of digital rectal examination and serum prostate specific antigen in the early detection of prostate cancer: Results of a multicenter clinical trial of 6630 men. J. Urol. 1994, 151, 1283–1290. [Google Scholar] [CrossRef]
- Harris, L.; Fritsche, H.; Mennel, R.; Norton, L.; Ravdin, P.; Taube, S.; Somerfield, M.R.; Hayes, D.F.; Bast, R.C. American Society of Clinical Oncology 2007 Update of Recommendations for the Use of Tumor Markers in Breast Cancer. J. Clin. Oncol. 2007, 25, 5287–5312. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science (N. Y.) 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef]
- Althuis, M.D.; Dozier, J.M.; Anderson, W.F.; Devesa, S.S.; Brinton, L.A. Global trends in breast cancer incidence and mortality 1973–1997. Int. J. Epidemiol. 2005, 34, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.H.F.; Coopey, S.B.; Freer, P.E.; Hughes, K.S. False-negative rate of combined mammography and ultrasound for women with palpable breast masses. Breast Cancer Res. Treat. 2015, 153, 699–702. [Google Scholar] [CrossRef]
- Polanski, M.; Anderson, N.L. A list of candidate cancer biomarkers for targeted proteomics. Biomark. Insights 2007, 1, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Hilvo, M.; Denkert, C.; Lehtinen, L.; Müller, B.; Brockmöller, S.; Seppänen-Laakso, T.; Budczies, J.; Bucher, E.; Yetukuri, L.; Castillo, S.; et al. Novel Theranostic Opportunities Offered by Characterization of Altered Membrane Lipid Metabolism in Breast Cancer Progression. Cancer Res. 2011, 71, 3236. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.H.; Fu, L.; Li, B.; Xie, H.L.; Zhang, X.; Chen, Y.; Qin, Y.; Wang, Y.; Zhang, S.; Huang, H.; et al. Distinguishing the serum metabolite profiles differences in breast cancer by gas chromatography mass spectrometry and random forest method. RSC Adv. 2015, 5, 58952–58958. [Google Scholar] [CrossRef]
- WHO Scientific Group on the Burden of Musculoskeletal Conditions at the Start of the New Millennium. The Burden of Musculoskeletal Conditions at the Start of the New Millennium; World Health Organization Technical Report Series; World Health Organization: Geneva, Switzerland, 2003; Volume 919, pp. 1–218. [Google Scholar]
- Peace, C.; Carr, A.; Loughlin, J. Recent advances in the genetic investigation of osteoarthritis. Trends Mol. Med. 2005, 11, 186–191. [Google Scholar]
- Wallace, I.J.; Worthington, S.; Felson, D.T.; Jurmain, R.D.; Wren, K.T.; Maijanen, H.; Woods, R.J.; Lieberman, D.E. Knee osteoarthritis has doubled in prevalence since the mid-20th century. Proc. Natl. Acad. Sci. USA 2017, 114, 9332–9336. [Google Scholar] [CrossRef]
- Loeser, R.F.; Collins, J.A.; Diekman, B.O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 412–420. [Google Scholar] [CrossRef]
- Zhang, Y.; Jordan, J.M. Epidemiology of osteoarthritis. Clin. Geriatr. Med. 2010, 26, 355–369. [Google Scholar] [CrossRef]
- Valdes, A.M.; Meulenbelt, I.; Chassaing, E.; Arden, N.K.; Bierma-Zeinstra, S.; Hart, D.; Hofman, A.; Karsdal, M.; Kloppenburg, M.; Kroon, H.M.; et al. Large scale meta-analysis of urinary C-terminal telopeptide, serum cartilage oligomeric protein and matrix metalloprotease degraded type II collagen and their role in prevalence, incidence and progression of osteoarthritis. Osteoarthr. Cartil. 2014, 22, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Likhodii, S.; Zhang, Y.; Aref-Eshghi, E.; Harper, P.E.; Randell, E.; Green, R.; Martin, G.; Furey, A.; Sun, G.; et al. Classification of osteoarthritis phenotypes by metabolomics analysis. BMJ Open 2014, 4, e006286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, H.; Zhang, Z.; Yang, F.; Chen, J. Serum metabolites as potential biomarkers for diagnosis of knee osteoarthritis. Dis. Mark. 2015, 2015, 684794. [Google Scholar] [CrossRef]
- Hu, T.; Oksanen, K.; Zhang, W.; Randell, E.; Furey, A.; Zhai, G. Analyzing feature importance for metabolomics using genetic programming. In Proceedings of the 21st European Conference (EuroGP 2018), Parma, Italy, 4–6 April 2018; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Peddinti, G.; Cobb, J.; Yengo, L.; Froguel, P.; Kravić, J.; Balkau, B.; Tuomi, T.; Aittokallio, T.; Groop, L. Early metabolic markers identify potential targets for the prevention of type 2 diabetes. Diabetologia 2017, 60, 1740–1750. [Google Scholar] [CrossRef] [PubMed]
- Gromski, P.S.; Muhamadali, H.; Ellis, D.I.; Xu, Y.; Correa, E.; Turner, M.L.; Goodacre, R. A tutorial review: Metabolomics and partial least squares-discriminant analysis—A marriage of convenience or a shotgun wedding. Anal. Chim. Acta 2015, 879, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, S.; Shah, S.L.; Marrie, T.J.; Slupsky, C.M. Analysis of metabolomic data using support vector machines. Anal. Chem. 2008, 80, 7562–7570. [Google Scholar] [CrossRef] [PubMed]
- Determan, C.E., Jr. Optimal algorithm for metabolomics classification and feature selection varies by dataset. Int. J. Biol. 2015, 7, 100–115. [Google Scholar]
- Brougham, D.F.; Ivanova, G.; Gottschalk, M.; Collins, D.M.; Eustace, A.J.; O’Connor, R.; Havel, J. Artificial neural networks for classification in metabolomic studies of whole cells using 1H nuclear magnetic resonance. J. Biomed. Biotechnol. 2001, 2011, I58094. [Google Scholar]
- Hall, L.M.; Hill, D.W.; Menikarachchi, L.C.; Chen, M.H.; Hall, L.H.; Grant, D.F. Optimizing artificial neural network models for metabolomics and systems biology: An example using HPLC retention index data. Bioanalysis 2015, 7, 939–955. [Google Scholar] [CrossRef]
- Kenny, L.C.; Dunn, W.B.; Ellis, D.I.; Myers, J.; Baker, P.N.; The GOPEC Consortium; Kell, D.B. Novel biomarkers for pre-eclampsia detected using metabolomics and machine learning. Metabolomics 2005, 1, 227–234. [Google Scholar] [CrossRef]
- Ghahramani, Z. Probabilistic machine learning and artificial intelligence. Nature 2015, 521, 452–459. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Statistical modelling: The two cultures. Stat. Sci. 2001, 16, 199–215. [Google Scholar] [CrossRef]
- Breiman, L. Bagging predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef]
- Dietterich, T.G. Ensemble methods in machine learning. Mult. Classif. Syst. 2000, 1857, 1–15. [Google Scholar]
- Schapire, R.E. The strength of weak learnability. Mach. Learn. 1990, 5, 197–227. [Google Scholar] [CrossRef]
- Freund, Y.; Schapire, R.E. A decision-theoretic generalization of on-line learning and an application to boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar] [CrossRef]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Anderson, J.A.; Rosenfeld, E. Neurocomputing: Foundations of Research; MIT Press: Cambridge, MA, USA, 1988. [Google Scholar]
- Wasserman, P.D. Neural Computing: Theory and Practice; Van Nostrand Reinhold: New York, NY, USA, 1989. [Google Scholar]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Silver, D.; Huang, A.; Maddison, C.J.; Guez, A.; Sifre, L.; van den Driessche, G.; Schrittwieser, J.; Antonoglou, I.; Panneershelvam, V.; Lanctot, M.; et al. Mastering the game of Go with deep neural networks and tree search. Nature 2016, 529, 484–489. [Google Scholar] [CrossRef]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet classification with deep convolutional neural networks. In Advances in Neural Information Processing Systems 25; MIT Press: Cambridge, MA, USA, 2012; pp. 1097–1105. [Google Scholar]
- Ackley, D.H.; Hinton, G.E.; Sejnowski, T.J. A learning algorithm for Boltzmann machines. Cogn. Sci. 1985, 9, 147–169. [Google Scholar] [CrossRef]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial networks. In Proceedings of the International Conference on Neural Information Processing Systems (NIPS), Montreal, QC, Canada, 8–13 December 2014; pp. 2672–2680. [Google Scholar]
- Holland, J. Adaptation in Natural and Artificial Systems; University of Michigan Press: Ann Arbor, MI, USA, 1975. [Google Scholar]
- Koza, J.R. Genetic Programming: On the Programming of Computers by Means of Natural Selection; MIT Press: Cambridge, MA, USA, 1992. [Google Scholar]
- Back, T.; Fogel, D.B.; Michalewicz, Z. Handbook of Evolutionary Computation; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Hu, T.; Banzhaf, W. Evolvability and speed of evolutionary algorithms in light of recent developments in Biology. J. Artif. Evol. Appl. 2010, 2010. [Google Scholar] [CrossRef]
- Goodacre, R.; Kell, D.B. Evolutionary computation for the interpretation of metabolomic data. In Metabolic Profiling: Its Role in Biomarker Discovery and Gene Function Analysis; Harrigan, G.G., Goodacre, H., Eds.; Kluwer Academic PUblishers: Dordrecht, The Netherlands, 2003; Chapter 13; pp. 239–256. [Google Scholar]
- Goodacre, R. Making sense of the metabolome using evolutionary computation: Seeing the wood with the trees. J. Exp. Bot. 2005, 56, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Bandyopadhyay, S.; Ray, S.S. Evolutionary computation in bioinformatics: A review. IEEE Trans. Syst. Man Cybern. Part C Appl. Rev. 2006, 36, 601–615. [Google Scholar] [CrossRef]
- Muni, D.P.; Pal, N.R.; Das, J. Genetic programming for simultaneous feature selection and classifier design. IEEE Trans. Syst. Man Cybern. Part B Cybern. 2006, 36, 106–117. [Google Scholar] [CrossRef]
- Hageman, J.A.; van den Berg, R.A.; Westerhuis, J.A.; van der Werf, M.J.; Smilde, A.K. Genetic algorithm based two-mode clustering of metabolomics data. Metabolomics 2008, 4, 141–149. [Google Scholar] [CrossRef]
- Devi, R.V.; Sathya, S.S.; Coumar, M.S. Evolutionary algorithms for de novo drug design—A survey. Appl. Soft Comput. 2015, 27, 543–552. [Google Scholar] [CrossRef]
- Banzhaf, W.; Nordin, P.; Keller, R.E.; Francone, F.D. Genetic Programming: An Introduction; Morgan Kaufmann: San Francisco, CA, USA, 1998. [Google Scholar]
- Brameier, M.F.; Banzhaf, W. Linear Genetic Programming; Springer: Berlin, Germany, 2007. [Google Scholar]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, G.; Likhodii, S.; Liu, M.; Aref-Eshghi, E.; Harper, P.E.; Martin, G.; Furey, A.; Green, R.; Randell, E.; et al. Metabolomic analysis of human plasma reveals that arginine is depleted in knee osteoarthritis patients. Osteoarthr. Cartil. 2016, 24, 827–834. [Google Scholar] [CrossRef]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef]
- Wilkins, J.M.; Trushina, E. Application of metabolomics in Alzheimer’s disease. Front. Neurol. 2018, 8, 719. [Google Scholar] [CrossRef]
- Nandania, J.; Peddinti, G.; Pessia, A.; Kokkonen, M.; Velagapudi, V. Validation and Automation of a High-Throughput Multitargeted Method for Semiquantification of Endogenous Metabolites from Different Biological Matrices Using Tandem Mass Spectrometry. Metabolites 2018, 8, 44. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Algorithm | Description | Examples |
|---|---|---|
| Logistic regression (LR) | Use a logistic function to fit a regression model for categorical outcome prediction. | [59] |
| Partial least squares-discriminant analysis (PLS-DA) | Find a linear subspace of high-dimensional explanatory variables to maximize the covariance between the input variables and the class label. | [60] |
| Support vector machine (SVM) | Use various similarity measures of training samples (also known as kernel functions) to perform linear or non-linear separation of two classes. | [61] |
| Random forest (RF) | Construct an ensemble of decision trees to classify training samples, as well as to assess the variable importance in the classification. | [25] |
| Gradient boosting machine (GBM) | Build an ensemble of decision trees in a step-wise fashion using boosting and gradient descent algorithms. | [62] |
| Artificial neural network (ANN) | Construct multi-layered networks of neurons to learn highly non-linear functions that map the explanatory variables to the class label. | [63,64] |
| Genetic programming (GP) | Use natural evolution mechanisms to automatically search for the most relevant features and classification models. | [7,65] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.Y.; Hu, T. Computational Methods for the Discovery of Metabolic Markers of Complex Traits. Metabolites 2019, 9, 66. https://doi.org/10.3390/metabo9040066
Lee MY, Hu T. Computational Methods for the Discovery of Metabolic Markers of Complex Traits. Metabolites. 2019; 9(4):66. https://doi.org/10.3390/metabo9040066
Chicago/Turabian StyleLee, Michael Y., and Ting Hu. 2019. "Computational Methods for the Discovery of Metabolic Markers of Complex Traits" Metabolites 9, no. 4: 66. https://doi.org/10.3390/metabo9040066
APA StyleLee, M. Y., & Hu, T. (2019). Computational Methods for the Discovery of Metabolic Markers of Complex Traits. Metabolites, 9(4), 66. https://doi.org/10.3390/metabo9040066