HILIC-Enabled 13C Metabolomics Strategies: Comparing Quantitative Precision and Spectral Accuracy of QTOF High- and QQQ Low-Resolution Mass Spectrometry



Abstract
1. Introduction
2. Results
2.1. Optimization of Chromatographic Conditions and ESI-MS Parameters
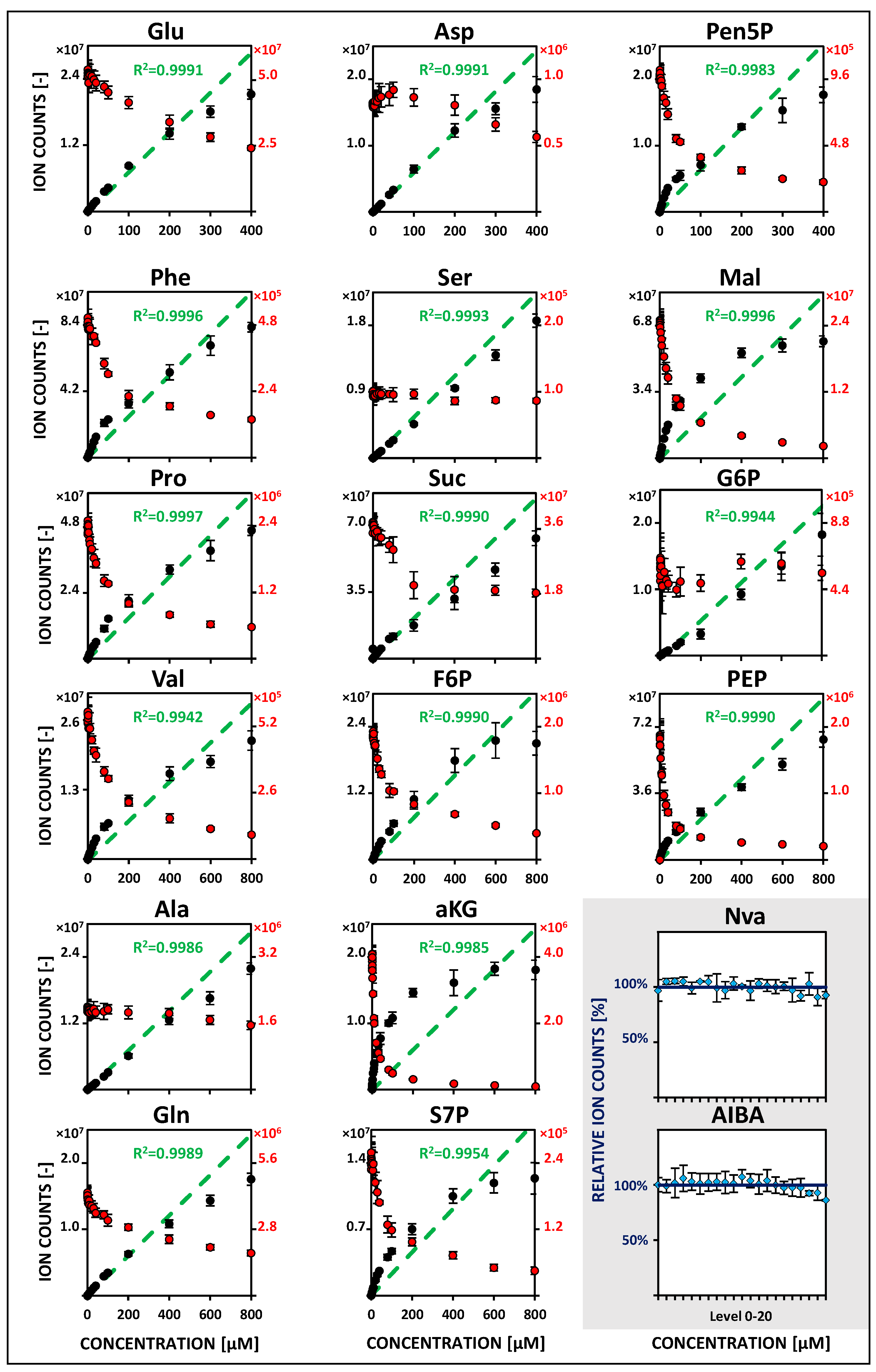
2.2. Metabolite-Specific Linearity Ranges and Sensitivities
2.3. Instrumental Precision, Retention Time Stabilities, and Methodical Detection Limits
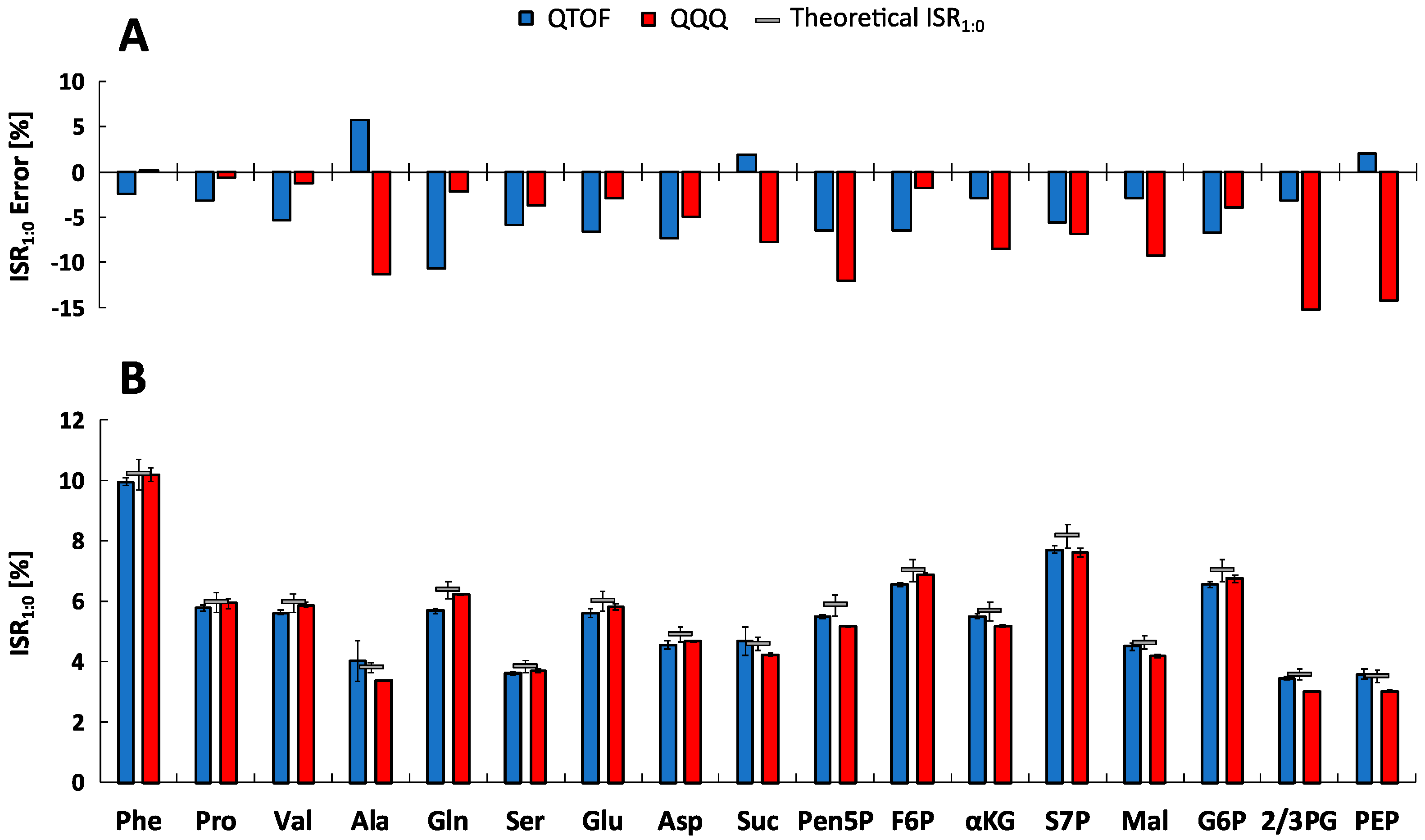
2.4. Isotope Dilution Mass Spectrometry (IDMS)
2.5. Metabolite-Specific Spectral Accuracies
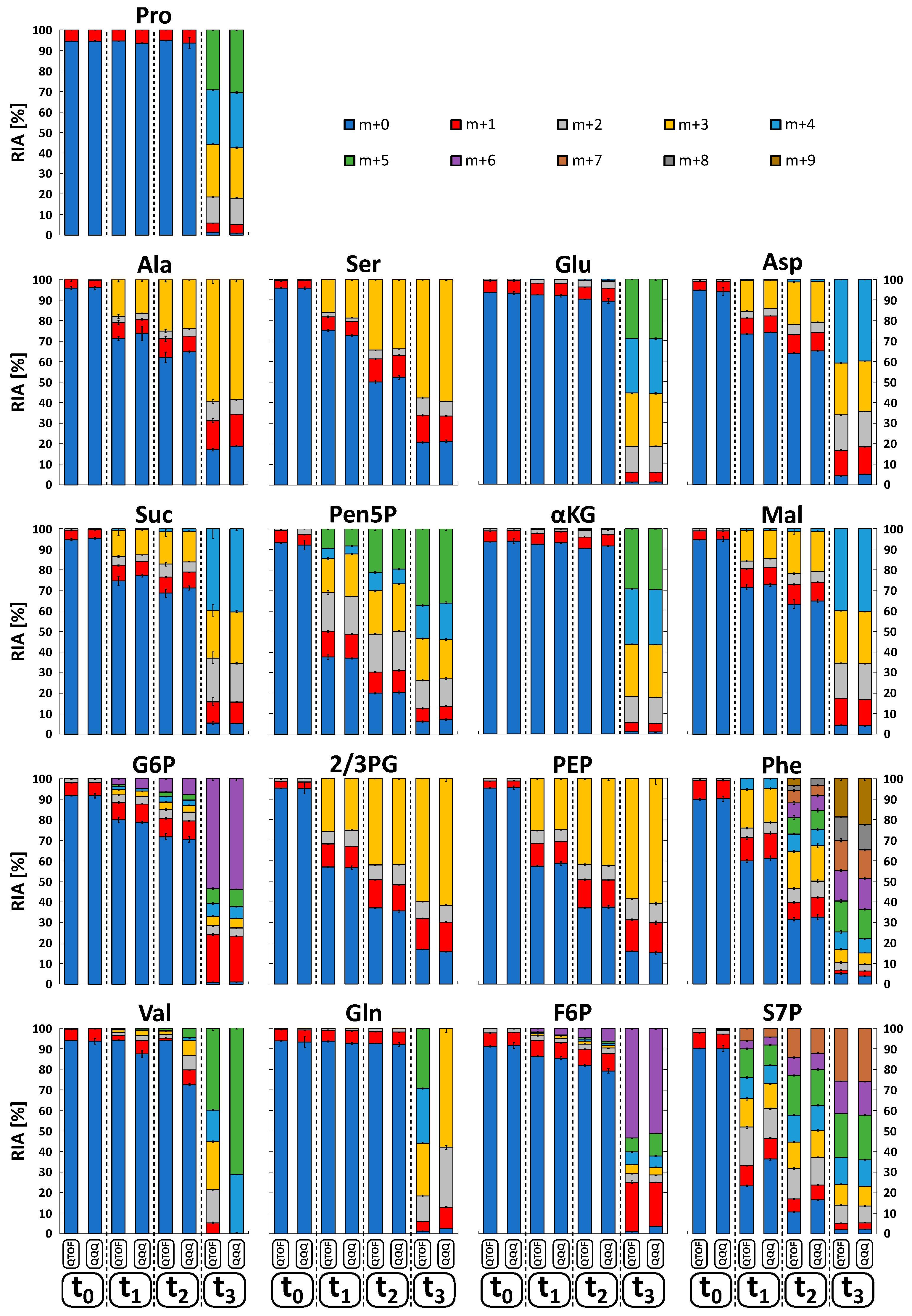
2.6. Isotopologue Analysis in Biological Sample Matrices
3. Discussion
4. Conclusions
5. Material and Methods
5.1. Analytical Chemicals
5.2. Strain and Seed Train
5.3. Preparation of Fully Labeled 13C-Metabolite Extracts from C. glutamicum
5.4. 13. C-labeling Experiments with C. glutamicum in Continuous Culture
5.5. Chromatographic Conditions
5.6. Data Acquisition for QQQ-MS/MS Studies
5.7. Data Acquisition for QTOF-HRMS Studies
5.8. Methodical Evaluation of ESI-MS-Based Signal Responsivities
5.9. Relative Quantification of Isotopic Labeling Dynamics
5.10. Statistical Analysis
Supplementary Materials
Author Contribution:
Funding
Acknowledgments
Conflicts of Interest
References
- Zamboni, N.; Fendt, S.-M.; Rühl, M.; Sauer, U. 13 C-based metabolic flux analysis. Nat. Protoc. 2009, 4, 878. [Google Scholar] [CrossRef] [PubMed]
- Nöh, K.; Droste, P.; Wiechert, W. Visual workflows for 13 C-metabolic flux analysis. Bioinformatics 2015, 31, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Niedenführ, S.; ten Pierick, A.; van Dam, P.T.N.; Suarez-Mendez, C.A.; Nöh, K.; Wahl, S.A. Natural isotope correction of MS/MS measurements for metabolomics and 13 C fluxomics. Biotechnol. Bioeng. 2015, 113, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, U.; Maier, K.; Reuss, M.; Mauch, K. Identification of metabolic fluxes in hepatic cells from transient 13 C-labeling experiments: Part II. Flux estimation. Biotechnol. Bioeng. 2007, 100, 355–370. [Google Scholar] [CrossRef]
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; DeBerardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A roadmap for interpreting 13C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015, 34, 189–201. [Google Scholar] [CrossRef]
- Dunn, W.B.; Ellis, D.I. Metabolomics: Current analytical platforms and methodologies. TrAC Trends Anal. Chem. 2005, 24, 285–294. [Google Scholar] [CrossRef]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef] [PubMed]
- Mashego, M.R.; Rumbold, K.; De Mey, M.; Vandamme, E.; Soetaert, W.; Heijnen, J.J. Microbial metabolomics: past, present and future methodologies. Biotechnol. Lett. 2007, 29, 1–16. [Google Scholar] [CrossRef]
- Purcaro, G.; Tranchida, P.Q.; Dugo, P.; Camera, E.L.; Bisignano, G.; Conte, L.; Mondello, L. Characterization of bacterial lipid profiles by using rapid sample preparation and fast comprehensive two-dimensional gas chromatography in combination with mass spectrometry. J. Sep. Sci. 2010, 33, 2334–2340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Halouska, S.; Schiaffo, C.E.; Sadykov, M.R.; Somerville, G.A.; Powers, R. NMR Analysis of a Stress Response Metabolic Signaling Network. J. Proteome Res. 2011, 10, 3743–3754. [Google Scholar] [CrossRef] [PubMed]
- Bean, H.D.; Dimandja, J.-M.D.; Hill, J.E. Bacterial volatile discovery using solid phase microextraction and comprehensive two-dimensional gas chromatography–time-of-flight mass spectrometry. J. Chromatogr. B 2012, 901, 41–46. [Google Scholar] [CrossRef]
- Choi, J.; Grossbach, M.T.; Antoniewicz, M.R. Measuring complete isotopomer distribution of aspartate using gas chromatography/tandem mass spectrometry. Anal. Chem. 2012, 84, 4628–4632. [Google Scholar] [CrossRef]
- Teleki, A.; Sánchez-Kopper, A.; Takors, R. Alkaline conditions in hydrophilic interaction liquid chromatography for intracellular metabolite quantification using tandem mass spectrometry. Anal. Biochem. 2015, 475, 4–13. [Google Scholar] [CrossRef]
- Al Kadhi, O.; Melchini, A.; Mithen, R.; Saha, S. Development of a LC-MS/MS Method for the Simultaneous Detection of Tricarboxylic Acid Cycle Intermediates in a Range of Biological Matrices. J. Anal. Methods Chem. 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Want, E.J.; Nordström, A.; Morita, H.; Siuzdak, G.; Nordstrm, A.; Morita, H.; Siuzdak, G.; Nordstro, A. From Exogenous to Endogenous: The Inevitable Imprint of Mass Spectrometry in Metabolomics. J. Proteome Res. 2007, 6, 459–468. [Google Scholar] [CrossRef]
- Koek, M.; Muilwijk, B.; Werf, M.J.; Hankemeier, T. Microbial metabolomics with gas chromatography mass spectrometry. Anal. Chem. 2006, 78, 1272–1281. [Google Scholar] [CrossRef]
- Preinerstorfer, B.; Schiesel, S.; Lämmerhofer, M.; Lindner, W. Metabolic profiling of intracellular metabolites in fermentation broths from β-lactam antibiotics production by liquid chromatography–tandem mass spectrometry methods. J. Chromatogr. A 2010, 1217, 312–328. [Google Scholar] [CrossRef]
- Dell’mour, M.; Jaitz, L.; Oburger, E.; Puschenreiter, M.; Koellensperger, G.; Hann, S. Hydrophilic interaction LC combined with electrospray MS for highly sensitive analysis of underivatized amino acids in rhizosphere research. J. Sep. Sci. 2010, 33, 911–922. [Google Scholar] [CrossRef]
- Yuan, M.; Breitkopf, S.B.; Yang, X.; Asara, J.M. A positive/negative ion–switching, targeted mass spectrometry–based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc. 2012, 7, 872–881. [Google Scholar] [CrossRef]
- Wiechert, W. 13C metabolic flux analysis. Metab. Eng. 2001, 3, 195–206. [Google Scholar] [CrossRef]
- Wang, Y.; Gu, M. The concept of spectral accuracy for MS. Anal. Chem. 2010, 82, 7055–7062. [Google Scholar] [CrossRef]
- Kind, T.; Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinform. 2007, 8, 1–20. [Google Scholar] [CrossRef]
- Hernández, F.; Ibáñez, M.; Sancho, J.V.; Pozo, Ó.J. Comparison of different mass spectrometric techniques combined with liquid chromatography for confirmation of pesticides in environmental water based on the use of identification points. Anal. Chem. 2004, 76, 4349–4357. [Google Scholar] [CrossRef]
- Lucini, L.; Pellegrino, R.; Cimino, N.; Kane, D.; Pretali, L. QqQ and Q-TOF liquid chromatography mass spectrometry direct aqueous analysis of herbicides and their metabolites in water. Int. J. Mass Spectrom. 2015, 392, 16–22. [Google Scholar] [CrossRef]
- Williamson, L.N.; Bartlett, M.G. Quantitative liquid chromatography/time-of-flight mass spectrometry. Biomed. Chromatogr. 2007, 21, 567–576. [Google Scholar] [CrossRef]
- Chen, S.; Li, M.; Zheng, G.; Wang, T.; Lin, J.; Wang, S.; Wang, X.; Chao, Q.; Cao, S.; Yang, Z.; et al. Metabolite profiling of 14 wuyi rock tea cultivars using UPLC-QTOF MS and UPLC-QqQ MS combined with chemometrics. Molecules 2018, 23, 104. [Google Scholar] [CrossRef]
- Chen, Y.J.; Liang, Z.T.; Zhu, Y.; Xie, G.Y.; Tian, M.; Zhao, Z.Z.; Qin, M.J. Tissue-specific metabolites profiling and quantitative analyses of flavonoids in the rhizome of Belamcanda chinensis by combining laser-microdissection with UHPLC-Q/TOF-MS and UHPLC–QqQ-MS. Talanta 2014, 130, 585–597. [Google Scholar] [CrossRef]
- Wells, G.; Prest, H.; Russ, C.W. Signal, Noise, and Detection Limits in Mass Spectrometry; Agilent Technologies: Wilmington, DE, USA, 2011. [Google Scholar]
- Somoano-Blanco, L.; Borda, M.; González Gago, A.; Rodríguez-González, P.; Garcia Alonso, J.I. Evaluation of the spectral accuracy of mass spectrometers using compounds containing Cl or Br atoms. J. Mass Spectrom. 2016, 51, 1036–1042. [Google Scholar] [CrossRef]
- Rühl, M.; Rupp, B.; Nöh, K.; Wiechert, W.; Sauer, U.; Zamboni, N. Collisional fragmentation of central carbon metabolites in LC-MS/MS increases precision of 13C metabolic flux analysis. Biotechnol. Bioeng. 2011, 109, 763–771. [Google Scholar] [CrossRef]
- Antoniewicz, M.R. Tandem mass spectrometry for measuring stable-isotope labeling. Curr. Opin. Biotechnol. 2013, 24, 48–53. [Google Scholar] [CrossRef]
- Holčapek, M.; Jirásko, R.; Lísa, M. Recent developments in liquid chromatography–mass spectrometry and related techniques. J. Chromatogr. A 2012, 1259, 3–15. [Google Scholar] [CrossRef]
- Luo, B.; Groenke, K.; Takors, R.; Wandrey, C.; Oldiges, M. Simultaneous determination of multiple intracellular metabolites in glycolysis, pentose phosphate pathway and tricarboxylic acid cycle by liquid chromatography—Mass spectrometry. J. Chromatogr. A 2007, 1147, 153–164. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Schug, K.A. The advantages of ESI-MS detection in conjunction with HILIC mode separations: Fundamentals and applications. J. Sep. Sci. 2008, 31, 1465–1480. [Google Scholar] [CrossRef]
- Spagou, K.; Tsoukali, H.; Raikos, N.; Gika, H.; Wilson, I.D.; Theodoridis, G. Hydrophilic interaction chromatography coupled to MS for metabonomic/metabolomic studies. J. Sep. Sci. 2010, 33, 716–727. [Google Scholar] [CrossRef]
- Alpert, A.J. Electrostatic Repulsion Hydrophilic Interaction Chromatography for Isocratic Separation of Charged Solutes and Selective Isolation of Phosphopeptides. Anal. Chem. 2008, 80, 62–76. [Google Scholar] [CrossRef]
- Annesley, T.M. Ion Suppression in Mass Spectrometry. Clin. Chem. 2003, 49, 1041–1044. [Google Scholar] [CrossRef]
- Schiesel, S.; Lämmerhofer, M.; Lindner, W. Multitarget quantitative metabolic profiling of hydrophilic metabolites in fermentation broths of β-lactam antibiotics production by HILIC-ESI-MS/MS. Anal. Bioanal. Chem. 2010, 396, 1655–1679. [Google Scholar] [CrossRef]
- Kiontke, A.; Oliveira-Birkmeier, A.; Opitz, A.; Birkemeyer, C. Electrospray Ionization Efficiency Is Dependent on Different Molecular Descriptors with Respect to Solvent pH and Instrumental Configuration. PLoS ONE 2016, 11, e0167502. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Mashego, M.R.; van Dam, J.C.; Proell, A.M.; Vinke, J.L.; Ras, C.; van Winden, W.A.; van Gulik, W.M.; Heijnen, J.J. Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly 13C-labeled cell extracts as internal standards. Anal. Biochem. 2005, 336, 164–171. [Google Scholar] [CrossRef]
- Kiefer, P.; Portais, J.-C.; Vorholt, J.A. Quantitative metabolome analysis using liquid chromatography–high-resolution mass spectrometry. Anal. Biochem. 2008, 382, 94–100. [Google Scholar] [CrossRef]
- Ciccimaro, E.; Blair, I.A. Stable-isotope dilution LC–MS for quantitative biomarker analysis. Bioanalysis 2010, 2, 311–341. [Google Scholar] [CrossRef]
- Vielhauer, O.; Zakhartsev, M.; Horn, T.; Takors, R.; Reuss, M. Simplified absolute metabolite quantification by gas chromatography–isotope dilution mass spectrometry on the basis of commercially available source material. J. Chromatogr. B 2011, 879, 3859–3870. [Google Scholar] [CrossRef]
- Erve, J.C.L.; Gu, M.; Wang, Y.; DeMaio, W.; Talaat, R.E. Spectral Accuracy of Molecular Ions in an LTQ/Orbitrap Mass Spectrometer and Implications for Elemental Composition Determination. J. Am. Soc. Mass Spectrom. 2009, 20, 2058–2069. [Google Scholar] [CrossRef]
- Xu, Y.; Heilier, J.F.; Madalinski, G.; Genin, E.; Ezan, E.; Tabet, J.C.; Junot, C. Evaluation of accurate mass and relative isotopic abundance measurements in the LTQ-Orbitrap mass spectrometer for further metabolomics database building. Anal. Chem. 2010, 82, 5490–5501. [Google Scholar] [CrossRef]
- Böcker, S.; Letzel, M.C.; Lipták, Z.; Pervukhin, A. SIRIUS: Decomposing isotope patterns for metabolite identification. Bioinformatics 2009, 25, 218–224. [Google Scholar] [CrossRef]
- Knolhoff, A.M.; Callahan, J.H.; Croley, T.R. Mass accuracy and isotopic abundance measurements for HR-MS instrumentation: Capabilities for non-targeted analyses. J. Am. Soc. Mass Spectrom. 2014, 25, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- González-Antuña, A.; Rodríguez-González, P.; García Alonso, J.I. Determination of the enrichment of isotopically labelled molecules by mass spectrometry. J. Mass Spectrom. 2014, 49, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Dauner, M.; Sauer, U. GC-MS analysis of amino acids rapidly provides rich information for isotopomer balancing. Biotechnol. Prog. 2000, 16, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Vanhaecke, F.; Degryse, P. Isotopic Analysis: Fundamentals and Applications Using ICP-MS; WILEY-VCH Verlag: Weinheim, Germany, 2012; ISBN 978-3-527-65050-7. [Google Scholar]
- Jochmann, M.A.; Schmidt, T.C. Compound-Specific Stable Isotope Analysis; The Royal Society of Chemistry: Cambridge, UK, 2012; ISBN 978-1-84973-157-7. [Google Scholar]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Accurate assessment of amino acid mass isotopomer distributions for metabolic flux analysis. Anal. Chem. 2007, 79, 7554–7559. [Google Scholar] [CrossRef]
- Teleki, A.; Rahnert, M.; Bungart, O.; Gann, B.; Ochrombel, I.; Takors, R. Robust identification of metabolic control for microbial l -methionine production following an easy-to-use puristic approach. Metab. Eng. 2017, 41, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, P.; Nicolas, C.; Letisse, F.; Portais, J.C. Determination of carbon labeling distribution of intracellular metabolites from single fragment ions by ion chromatography tandem mass spectrometry. Anal. Biochem. 2007, 360, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbour Laboratory Press: New York, NY, USA, 2001; ISBN 9780879695767. [Google Scholar]
- Buchholz, J.; Graf, M.; Freund, A.; Busche, T.; Kalinowski, J.; Blombach, B.; Takors, R. CO2/HCO3− perturbations of simulated large scale gradients in a scale-down device cause fast transcriptional responses in Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2014, 98, 8563–8572. [Google Scholar] [CrossRef] [PubMed]
- De Koning, W.; Dam, K. van A method for the determination of changes of glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral pH. Anal. Biochem. 1992, 204, 118–123. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Linearity and Sensitivity | Precision and Detection Limit | Performance | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name | 12C-Calibration | U13C-IDMS | 12C-Calibration | U13C-IDMS | LC-MS Parameters | ||||||||
| Abbr. | Min (nM) | Max (µM) | Slope (Cts/nM) | R2 (-) | Min (nM) | Max (nM) | R2 (-) | MV RSD (%) | MDL (nM) | MV RSD (%) | MDL (nM) | Retention tR (min) | ESI Polarity |
| Phe | 10 | 100 | 1424 | 0.9975 | 10 | 800 | 0.9954 | 1.94 | 3.5 | 1.94 | 2.8 | 10.12 ± 0.04 | [+] |
| Pro | 50 | 100 | 1346 | 0.9938 | 10 | 800 | 0.9948 | 4.72 | 15.5 | 1.75 | 4.3 | 12.47 ± 0.01 | [+] |
| Val | 200 | 100 | 703 | 0.9955 | 200 | 800 | 0.9986 | 1.87 | 17.3 | 2.09 | 35.2 | 12.66 ± 0.01 | [+] |
| Ala | 50 | 400 | 244 | 0.9905 | 50 | 800 | 0.9991 | 2.13 | 1.4 | 2.34 | 6.6 | 15.86 ± 0.01 | [+] |
| Gln | 50 | 800 | 144 | 0.9919 | 50 | 800 | 0.9997 | 2.96 | 9.6 | 1.48 | 5.0 | 17.16 ± 0.01 | [+] |
| Ser | 100 | 600 | 120 | 0.9932 | 100 | 800 | 0.9999 | 2.65 | 60.9 | 2.74 | 64.5 | 17.86 ± 0.01 | [+] |
| Glu | 50 | 400 | 189 | 0.9983 | 50 | 400 | 0.9999 | 2.18 | 12.2 | 1.54 | 7.3 | 19.7 ± 0.01 | [+] |
| Asp | 25 | 400 | 101 | 0.9994 | 25 | 400 | 0.9998 | 3.02 | 3.1 | 2.40 | 2.7 | 20.17 ± 0.01 | [+] |
| Suc | 100 | 800 | 18 | 0.9914 | 100 | 800 | 0.9995 | 4.06 | 55.1 | 3.40 | 65.9 | 20.81 ± 0.04 | [−] |
| Pen5P * | 25 | 20 | 66 | 0.9949 | 25 | 400 | 0.9998 | 2.30 | 6.3 | 2.40 | 6.1 | 21.4 ± 0.05 | [−] |
| F6P | 50 | 400 | 38 | 0.9907 | 50 | 800 | 0.9940 | 2.01 | 5.1 | 2.36 | 5.9 | 21.97 ± 0.03 | [−] |
| αKG | 50 | 10 | 35 | 0.9903 | 50 | 800 | 0.9996 | 2.14 | 11.8 | 2.43 | 13.4 | 22.11 ± 0.07 | [−] |
| S7P | 50 | 600 | 35 | 0.9914 | 50 | 800 | 0.9996 | 3.13 | 17.4 | 3.34 | 12.6 | 22.34 ± 0.04 | [−] |
| Mal | 50 | 40 | 93 | 0.9946 | 50 | 800 | 0.9990 | 3.71 | 19.9 | 2.70 | 13.2 | 22.55 ± 0.08 | [−] |
| G6P | 50 | 800 | 30 | 0.9988 | 50 | 800 | 0.9997 | 2.59 | 4.2 | 2.59 | 6.8 | 23.04 ± 0.03 | [−] |
| PEP | 50 | 4 | 139 | 0.9975 | 50 | 800 | 0.9998 | 1.98 | 9.7 | 2.05 | 12.5 | 24.72 ± 0.10 | [−] |
| Metabolite | Linearity and Sensitivity | Precision and Detection Limit | Performance | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name | 12C-Calibration | U13C-IDMS | 12C-Calibration | U13C-IDMS | LC-MS Parameters | ||||||||
| Abbr. | Min (nM) | Max (µM) | Slope (Cts/nM) | R2 (-) | Min (nM) | Max (nM) | R2 (-) | MV RSD (%) | MDL (nM) | MV RSD (%) | MDL (nM) | Retention tR (min) | ESI Polarity |
| Phe | 50 | 40 | 329 | 0.9995 | 50 | 800 | 0.9996 | 5.45 | 28.1 | 5.75 | 41.7 | 11.95 ± 0.03 | [+] |
| Pro | 50 | 100 | 142 | 0.9943 | 50 | 800 | 0.9997 | 8.73 | 39.3 | 7.17 | 43.3 | 13.99 ± 0.02 | [+] |
| Val | 400 | 40 | 96 | 0.9949 | 400 | 800 | 0.9942 | 6.08 | 75.5 | 6.53 | 49.3 | 14.15 ± 0.02 | [+] |
| Ala | 50 | 800 | 28 | 0.9967 | 50 | 800 | 0.9986 | 7.45 | 33.7 | 5.50 | 27.1 | 16.88 ± 0.02 | [−] |
| Gln | 50 | 400 | 28 | 0.9900 | 50 | 800 | 0.9989 | 6.18 | 19.9 | 4.54 | 31.1 | 17.94 ± 0.02 | [−] |
| Ser | 100 | 800 | 23 | 0.9999 | 100 | 800 | 0.9993 | 9.47 | 75.2 | 6.81 | 88.8 | 18.52 ± 0.02 | [−] |
| Glu | 50 | 200 | 73 | 0.9922 | 50 | 400 | 0.9991 | 5.78 | 22.7 | 3.71 | 17.6 | 19.97 ± 0.01 | [−] |
| Asp | 200 | 200 | 62 | 0.9991 | 200 | 400 | 0.9991 | 9.27 | 159.2 | 4.51 | 93.5 | 20.36 ± 0.02 | [−] |
| Suc | 200 | 800 | 78 | 0.9950 | 200 | 800 | 0.9990 | 8.51 | 151.7 | 4.73 | 150.1 | 21.05 ± 0.04 | [−] |
| Pen5P * | 200 | 15 | 202 | 0.9938 | 200 | 400 | 0.9983 | 6.48 | 89.3 | 7.15 | 105.0 | 21.49 ± 0.04 | [−] |
| F6P | 200 | 40 | 84 | 0.9923 | 200 | 800 | 0.9990 | 9.27 | 176.3 | 7.35 | 139.9 | 21.94 ± 0.04 | [−] |
| αKG | 50 | 2 | 876 | 0.9969 | 50 | 800 | 0.9985 | 6.68 | 5.7 | 7.07 | 12.7 | 22.18 ± 0.06 | [−] |
| S7P | 100 | 30 | 86 | 0.9943 | 100 | 800 | 0.9954 | 8.75 | 32.4 | 4.98 | 23.4 | 22.30 ± 0.05 | [−] |
| Mal | 50 | 30 | 509 | 0.9940 | 50 | 800 | 0.9996 | 5.13 | 13.5 | 4.00 | 18.5 | 22.57 ± 0.06 | [−] |
| G6P | 200 | 800 | 23 | 0.9964 | 200 | 800 | 0.9944 | 17.70 | 116.8 | 11.05 | 93.2 | 22.89 ± 0.03 | [−] |
| PEP | 100 | 10 | 448 | 0.9905 | 100 | 800 | 0.9990 | 7.66 | 71.8 | 4.60 | 31.7 | 24.54 ± 0.07 | [−] |
| Metabolite | 12C/U13C-Isotopic Purity (%) | U13C-Concentration (µM) | |||
|---|---|---|---|---|---|
| Abbr. | Name | QQQ | QTOF | QQQ | QTOF |
| Phe | l-Phenylalanine | 1.64 ± 0.09 | 1.32 ± 0.47 | 1.18 ± 0.01 | 1.33 ± 0.07 |
| Pro | l-Proline | 0.51 ± 0.13 | 0.35 ± 0.05 | 9.02 ± 0.16 | 10.67 ± 1.00 |
| Val | l-Valine | <MDL | <MDL | 3.82 ± 0.07 | 3.88 ± 0.37 |
| Ala | l-Alanine | 0.13 ± 0.01 | 0.15 ± 0.02 | 64.85 ± 1.40 | 62.75 ± 1.28 |
| Gln | l-Glutamine | 0.03 ± 0.00 | 0.01 ± 0.01 | 98.37 ± 1.55 | 92.09 ± 5.87 |
| Ser | l-Serine | 5.53 ± 0.49 | 3.25 ± 4.63 | 3.93 ± 0.03 | 4.04 ± 0.10 |
| Glu | l-Glutamate | 0.03 ± 0.00 | 0.04 ± 0.01 | 488.95 ± 7.62 | 460.00 ± 27.48 |
| Asp | l-Aspartate | 0.42 ± 0.03 | 0.08 ± 0.09 | 13.99 ± 0.18 | 14.55 ± 0.62 |
| Suc | Succinate | <MDL | <MDL | 251.74 ± 4.06 | 240.24 ± 9.44 |
| Pen5P * | Pentose-5-phosphate * | 1.37 ± 0.16 | 2.49 ± 0.24 | 4.21 ± 0.08 | 3.96 ± 0.09 |
| F6P | d-Fructose-6-phosphate | 0.31 ± 0.02 | 0.80 ± 0.12 | 13.50 ± 0.28 | 15.46 ± 0.41 |
| αKG | α-Ketoglutarate | 0.89 ± 0.05 | 1.93 ± 0.09 | 3.71 ± 0.04 | 4.14 ± 0.17 |
| S7P | d-Sedoheptulose 7-phosphate | 2.04 ± 0.12 | 3.57 ± 0.83 | 2.35 ± 0.02 | 2.34 ± 0.09 |
| Mal | l-Malate | <MDL | <MDL | 36.82 ± 0.47 | 32.74 ± 0.56 |
| G6P | d-Glucose-6-phosphate | 0.26 ± 0.02 | 0.27 ± 0.46 | 24.24 ± 0.37 | 24.76 ± 3.66 |
| PEP | 2-Phosphoenolpyruvate | 1.42 ± 0.07 | 2.00 ± 0.24 | 2.80 ± 0.08 | 2.75 ± 0.09 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feith, A.; Teleki, A.; Graf, M.; Favilli, L.; Takors, R. HILIC-Enabled 13C Metabolomics Strategies: Comparing Quantitative Precision and Spectral Accuracy of QTOF High- and QQQ Low-Resolution Mass Spectrometry. Metabolites 2019, 9, 63. https://doi.org/10.3390/metabo9040063
Feith A, Teleki A, Graf M, Favilli L, Takors R. HILIC-Enabled 13C Metabolomics Strategies: Comparing Quantitative Precision and Spectral Accuracy of QTOF High- and QQQ Low-Resolution Mass Spectrometry. Metabolites. 2019; 9(4):63. https://doi.org/10.3390/metabo9040063
Chicago/Turabian StyleFeith, André, Attila Teleki, Michaela Graf, Lorenzo Favilli, and Ralf Takors. 2019. "HILIC-Enabled 13C Metabolomics Strategies: Comparing Quantitative Precision and Spectral Accuracy of QTOF High- and QQQ Low-Resolution Mass Spectrometry" Metabolites 9, no. 4: 63. https://doi.org/10.3390/metabo9040063
APA StyleFeith, A., Teleki, A., Graf, M., Favilli, L., & Takors, R. (2019). HILIC-Enabled 13C Metabolomics Strategies: Comparing Quantitative Precision and Spectral Accuracy of QTOF High- and QQQ Low-Resolution Mass Spectrometry. Metabolites, 9(4), 63. https://doi.org/10.3390/metabo9040063