MetaboAnalystR 2.0: From Raw Spectra to Biological Insights



Abstract

1. Introduction
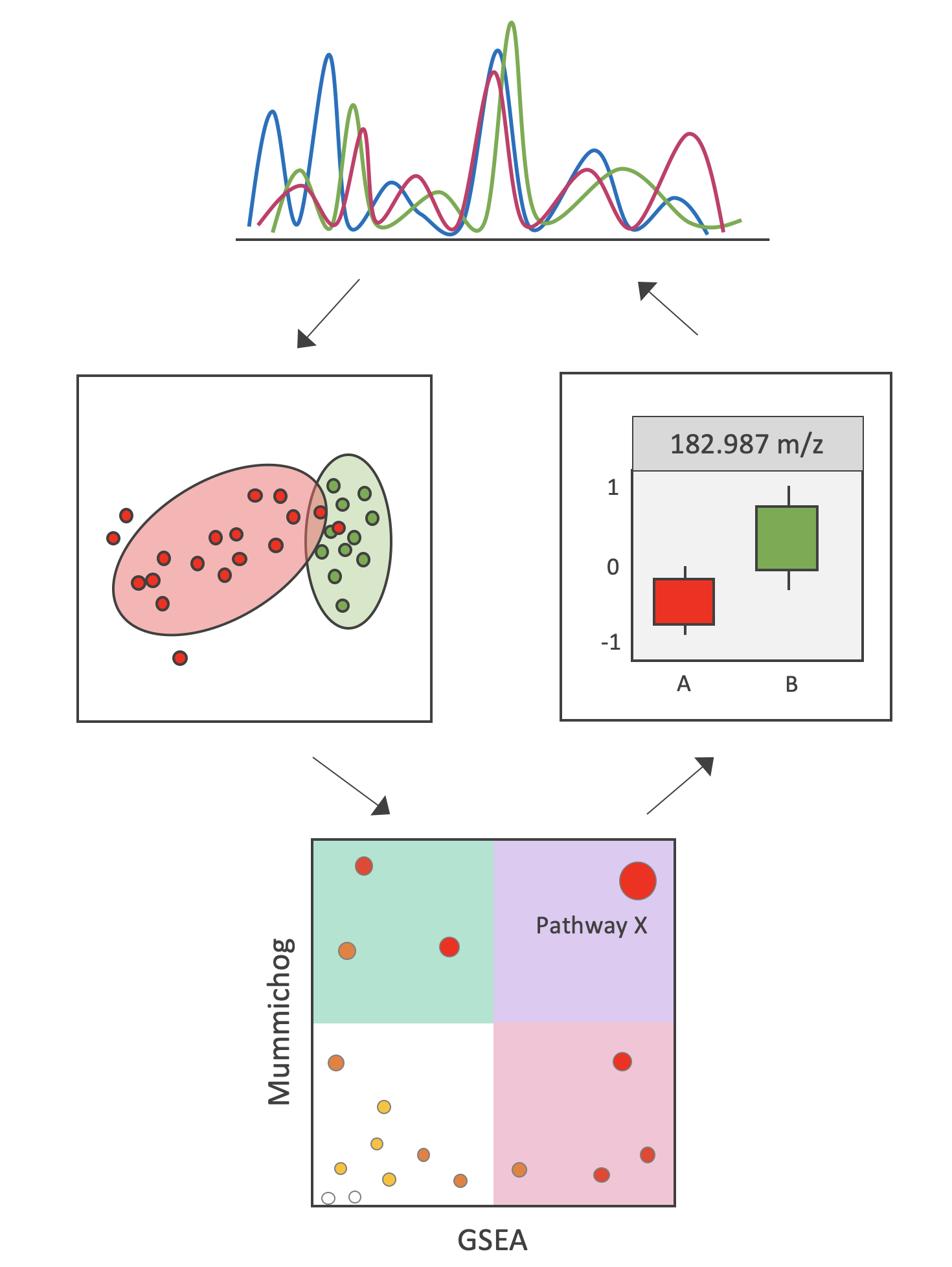
2. Results
2.1. Benchmark Case Study
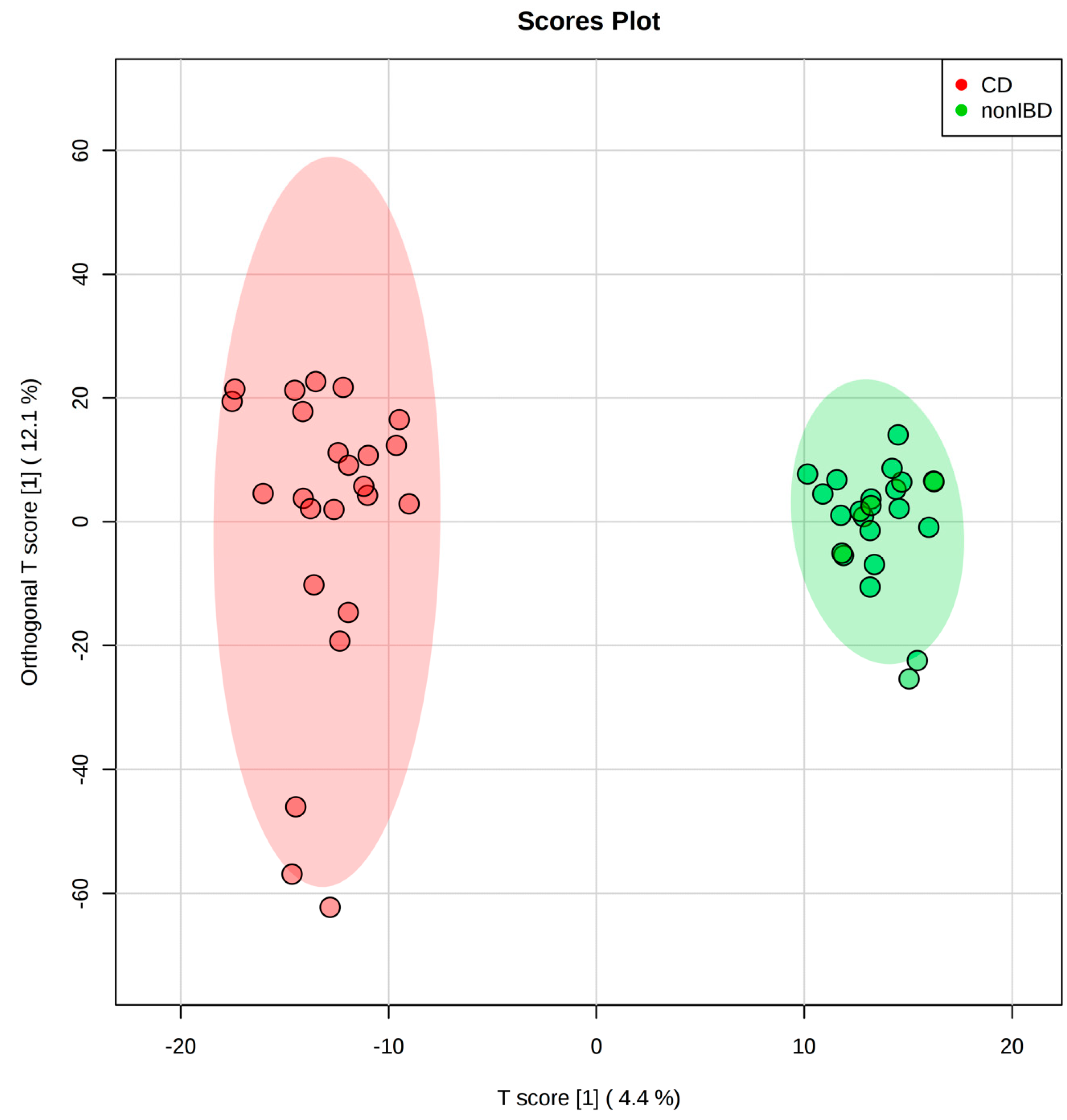
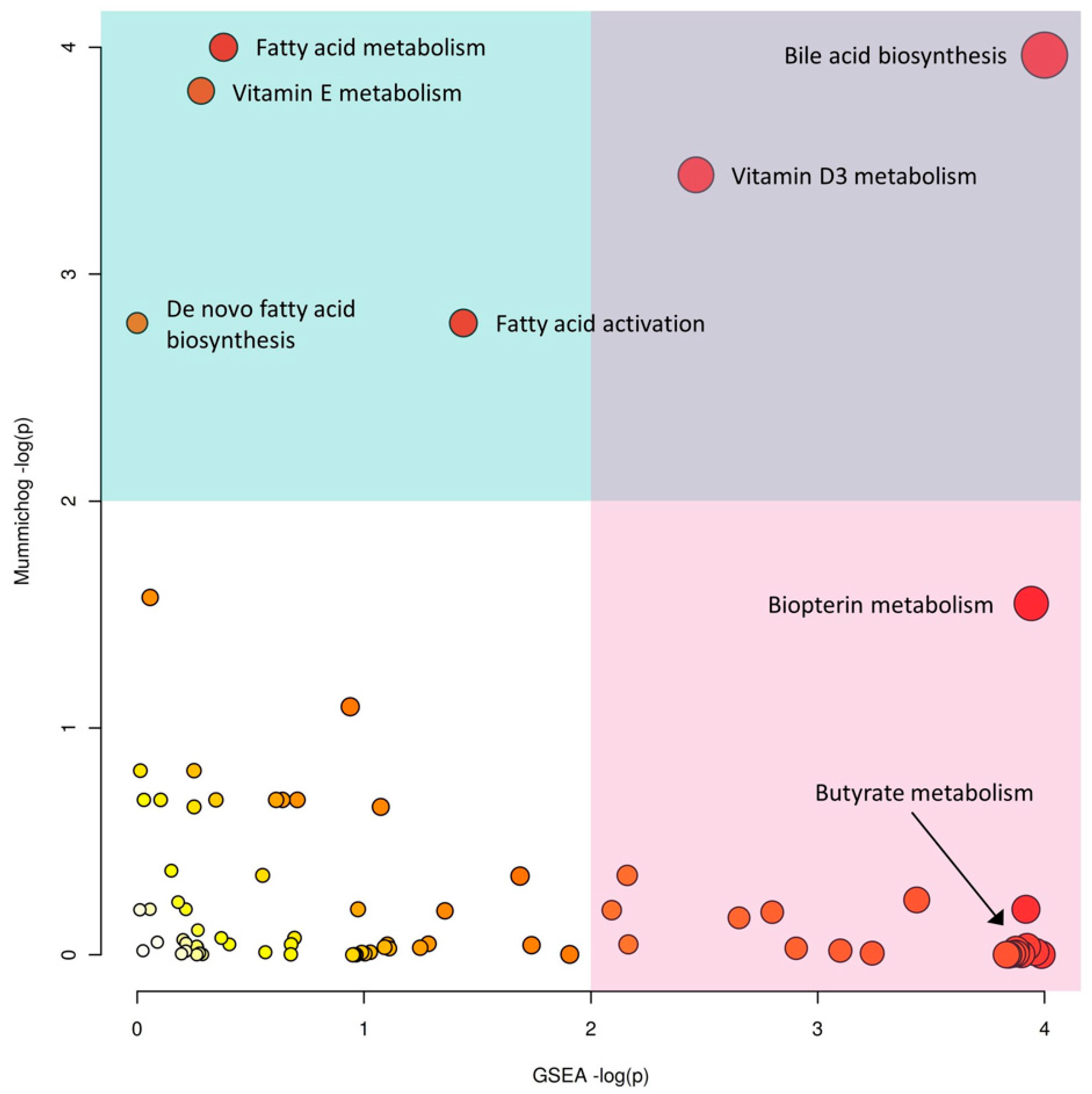
2.2. IBD Case Study
3. Discussion
4. Conclusions
5. Materials and Methods
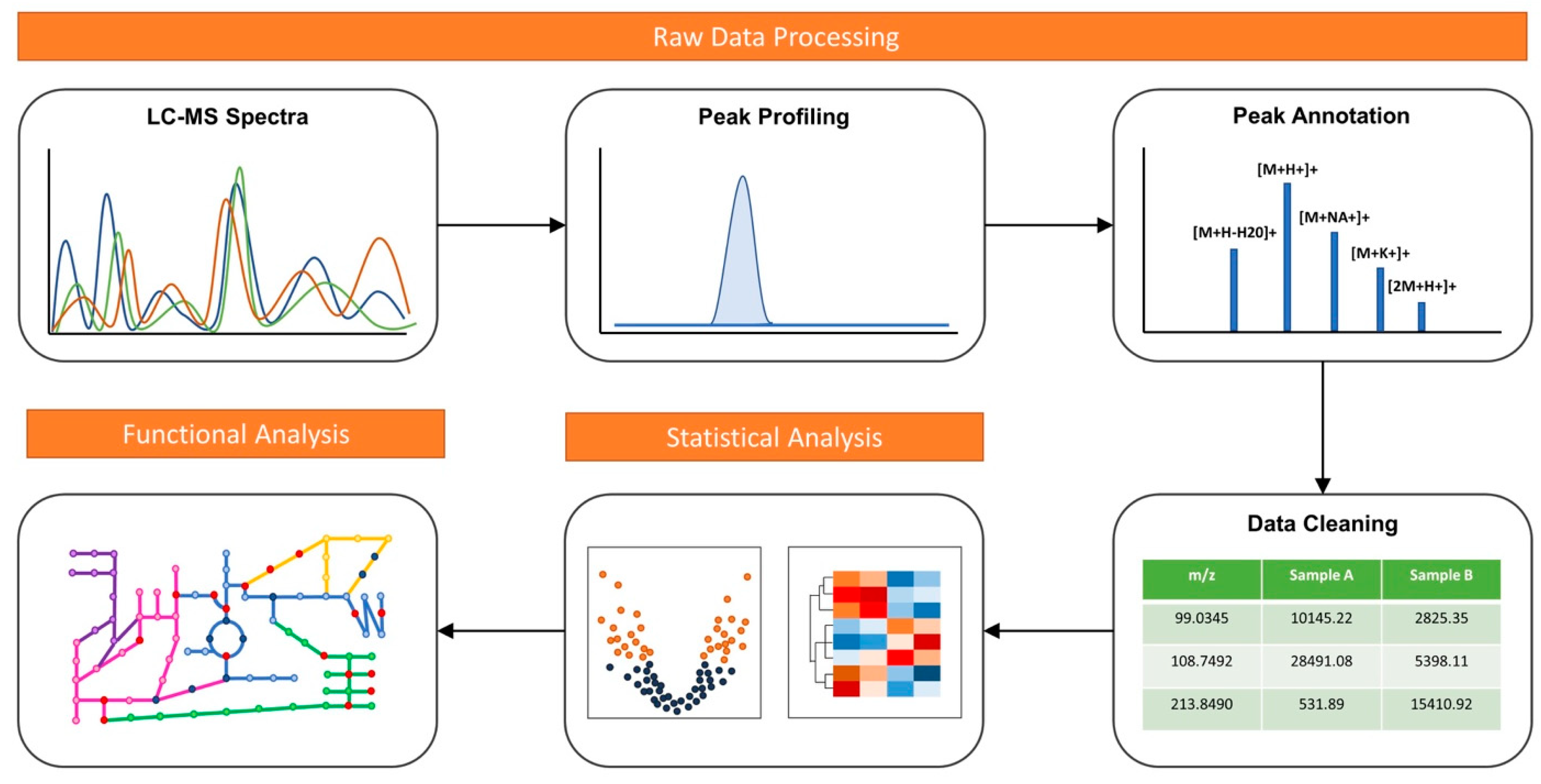
5.1. Spectral Processing
5.2. Prediction of Pathway Activities
5.3. Benchmark Case Studies
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beger, R.D.; Dunn, W.; Schmidt, M.A.; Gross, S.S.; Kirwan, J.A.; Cascante, M.; Brennan, L.; Wishart, D.S.; Oresic, M.; Hankemeier, T. Metabolomics enables precision medicine: “A white paper, community perspective”. Metabolomics 2016, 12, 149. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell biol. 2016, 17, 451. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. In Functional Genomics; Springer: Berlin/Heidelberg, Germany, 2002; pp. 155–171. [Google Scholar]
- Nash, W.J.; Dunn, W.B. From mass to metabolite in human untargeted metabolomics: Recent advances in annotation of metabolites applying liquid chromatography-mass spectrometry data. TrAC Trends Anal. Chem. 2018. [Google Scholar] [CrossRef]
- Uppal, K.; Walker, D.I.; Liu, K.; Li, S.; Go, Y.-M.; Jones, D.P. Computational metabolomics: A framework for the million metabolome. Chem. Res. Toxicol. 2016, 29, 1956–1975. [Google Scholar] [CrossRef]
- Forsberg, E.M.; Huan, T.; Rinehart, D.; Benton, H.P.; Warth, B.; Hilmers, B.; Siuzdak, G. Data processing, multi-omic pathway mapping, and metabolite activity analysis using XCMS Online. Nat. Protoc. 2018, 13, 633. [Google Scholar] [CrossRef] [PubMed]
- Giacomoni, F.; Le Corguillé, G.; Monsoor, M.; Landi, M.; Pericard, P.; Pétéra, M.; Duperier, C.; Tremblay-Franco, M.; Martin, J.-F.; Jacob, D. Workflow4Metabolomics: A collaborative research infrastructure for computational metabolomics. Bioinformatics 2014, 31, 1493–1495. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Rost, H.L.; Sachsenberg, T.; Aiche, S.; Bielow, C.; Weisser, H.; Aicheler, F.; Andreotti, S.; Ehrlich, H.C.; Gutenbrunner, P.; Kenar, E.; et al. OpenMS: A flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 2016, 13, 741–748. [Google Scholar] [CrossRef]
- Fernández-Albert, F.; Llorach, R.; Andrés-Lacueva, C.; Perera, A. An R package to analyse LC/MS metabolomic data: MAIT (Metabolite Automatic Identification Toolkit). Bioinformatics 2014, 30, 1937–1939. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Mei, Z.; Zeng, C.; Liu, S. metaX: A flexible and comprehensive software for processing metabolomics data. BMC Bioinform. 2017, 18, 183. [Google Scholar] [CrossRef] [PubMed]
- Xia, J. Computational Strategies for Biological Interpretation of Metabolomics Data. Adv. Exp. Med. Biol. 2017, 965, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Gardinassi, L.G.; Xia, J.; Safo, S.E.; Li, S. Bioinformatics Tools for the Interpretation of Metabolomics Data. Curr. Pharmacol. Rep. 2017, 3, 374–383. [Google Scholar] [CrossRef]
- Kind, T.; Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinform. 2007, 8, 105. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010, 38, W71–W77. [Google Scholar] [CrossRef]
- Li, S.; Park, Y.; Duraisingham, S.; Strobel, F.H.; Khan, N.; Soltow, Q.A.; Jones, D.P.; Pulendran, B. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 2013, 9, e1003123. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef]
- Xia, J.; Mandal, R.; Sinelnikov, I.V.; Broadhurst, D.; Wishart, D.S. MetaboAnalyst 2.0—A comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012, 40, W127–W133. [Google Scholar] [CrossRef]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef]
- Chong, J.; Xia, J. MetaboAnalystR: An R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 2018, 34, 4313–4314. [Google Scholar] [CrossRef]
- Li, Z.; Lu, Y.; Guo, Y.; Cao, H.; Wang, Q.; Shui, W. Comprehensive evaluation of untargeted metabolomics data processing software in feature detection, quantification and discriminating marker selection. Anal. Chim. Acta 2018, 1029, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011, 40, D109–D114. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.-A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef]
- Hofmann, A.; Hagey, L. Bile acids: Chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell. Mol. Life Sci. 2008, 65, 2461–2483. [Google Scholar] [CrossRef]
- Limketkai, B.N.; Mullin, G.E.; Limsui, D.; Parian, A.M. Role of vitamin D in inflammatory bowel disease. Nutr. Clin. Pract. 2017, 32, 337–345. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed]
- Tautenhahn, R.; Boettcher, C.; Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinform. 2008, 9, 504. [Google Scholar] [CrossRef] [PubMed]
- Benton, H.P.; Want, E.J.; Ebbels, T.M. Correction of mass calibration gaps in liquid chromatography-mass spectrometry metabolomics data. Bioinformatics 2010, 26, 2488–2489. [Google Scholar] [CrossRef]
- Kuhl, C.; Tautenhahn, R.; Bottcher, C.; Larson, T.R.; Neumann, S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 2011, 84, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sergushichev, A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. BioRxiv 2016. [Google Scholar] [CrossRef]
- Consortium, I.H.i.R.N. The Integrative Human Microbiome Project: Dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe 2014, 16, 276. [Google Scholar]
- Holman, J.D.; Tabb, D.L.; Mallick, P. Employing ProteoWizard to convert raw mass spectrometry data. Curr. Protoc. Bioinform. 2014, 46, 13.24.1–13.24.9. [Google Scholar]
- Di Guida, R.; Engel, J.; Allwood, J.W.; Weber, R.J.; Jones, M.R.; Sommer, U.; Viant, M.R.; Dunn, W.B. Non-targeted UHPLC-MS metabolomic data processing methods: A comparative investigation of normalisation, missing value imputation, transformation and scaling. Metabolomics 2016, 12, 93. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Features Detected | True Features | |||
|---|---|---|---|---|---|
| Total | Accurately Quantified | Discriminating | |||
| Li et al. 2018 [25] | Targeted | - | 836 | 836 | - |
| Untargeted (XCMS Online) | 35215 | 820 | 731 | 45 | |
| MetaboAnalystR 2.0 | Untargeted | 21013 | 732 | 632 | 45 |
| Mummichog | GSEA | ||||
|---|---|---|---|---|---|
| Pathway Name | Compound Hits * | p-Value | Pathway Name | Compound Hits | p-Value |
| Bile acid biosynthesis | 29/52 | 0.00282 | Bile acid biosynthesis | 52 | 0.001761 |
| Vitamin E metabolism | 20/33 | 0.00356 | Androgen and estrogen biosynthesis and metabolism | 10 | 0.01465 |
| Fatty acid metabolism | 9/11 | 0.00268 | Squalene and cholesterol biosynthesis | 7 | 0.02214 |
| Vitamin D3 metabolism | 8/10 | 0.00616 | Biopterin metabolism | 14 | 0.07806 |
| Fatty acid activation | 10/15 | 0.01620 | Butyrate metabolism | 11 | 0.08318 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chong, J.; Yamamoto, M.; Xia, J. MetaboAnalystR 2.0: From Raw Spectra to Biological Insights. Metabolites 2019, 9, 57. https://doi.org/10.3390/metabo9030057
Chong J, Yamamoto M, Xia J. MetaboAnalystR 2.0: From Raw Spectra to Biological Insights. Metabolites. 2019; 9(3):57. https://doi.org/10.3390/metabo9030057
Chicago/Turabian StyleChong, Jasmine, Mai Yamamoto, and Jianguo Xia. 2019. "MetaboAnalystR 2.0: From Raw Spectra to Biological Insights" Metabolites 9, no. 3: 57. https://doi.org/10.3390/metabo9030057
APA StyleChong, J., Yamamoto, M., & Xia, J. (2019). MetaboAnalystR 2.0: From Raw Spectra to Biological Insights. Metabolites, 9(3), 57. https://doi.org/10.3390/metabo9030057