Consistency, Inconsistency, and Ambiguity of Metabolite Names in Biochemical Databases Used for Genome-Scale Metabolic Modelling

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results
- Identifier (ID): Identifiers are strings of alpha-numeric characters used to identify uniquely a metabolite or a reaction in a database. Examples are C00001 in KEGG or ATPM in BiGG.
- Name: Here we use name to refer not only to the chemical name, but also to the set of aliases, synonyms, and abbreviations that are often included in a database as other names of the compound. For instance, the KEGG ID C00001 is associated with the name ‘water’.
- Multiplicity: describes the case on which a single ID is linked to multiple names. For instance, the KEGG ID C00001 is associated with the names ‘water’ and ‘H20’; therefore, we state that this ID has a multiplicity of 2.
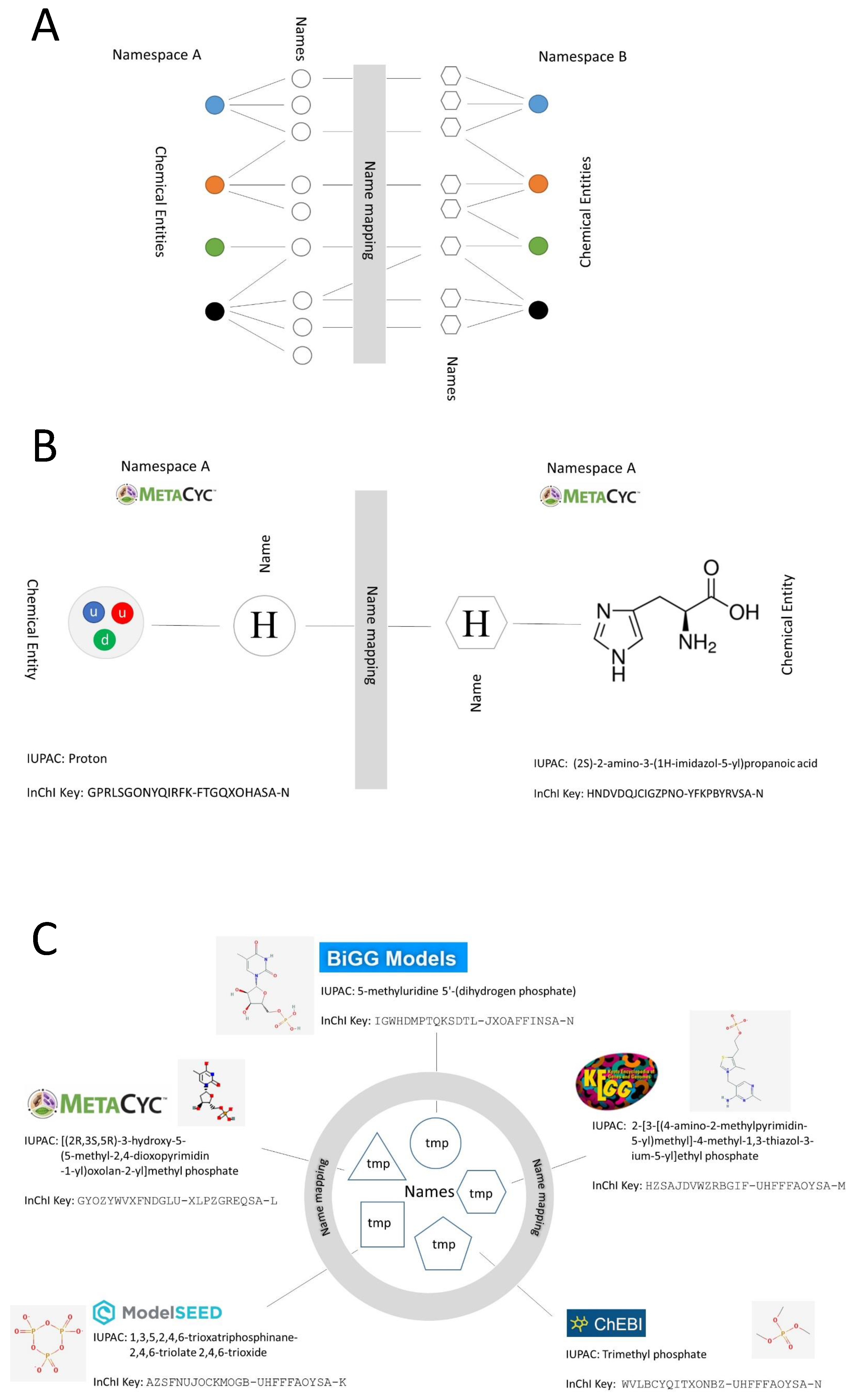
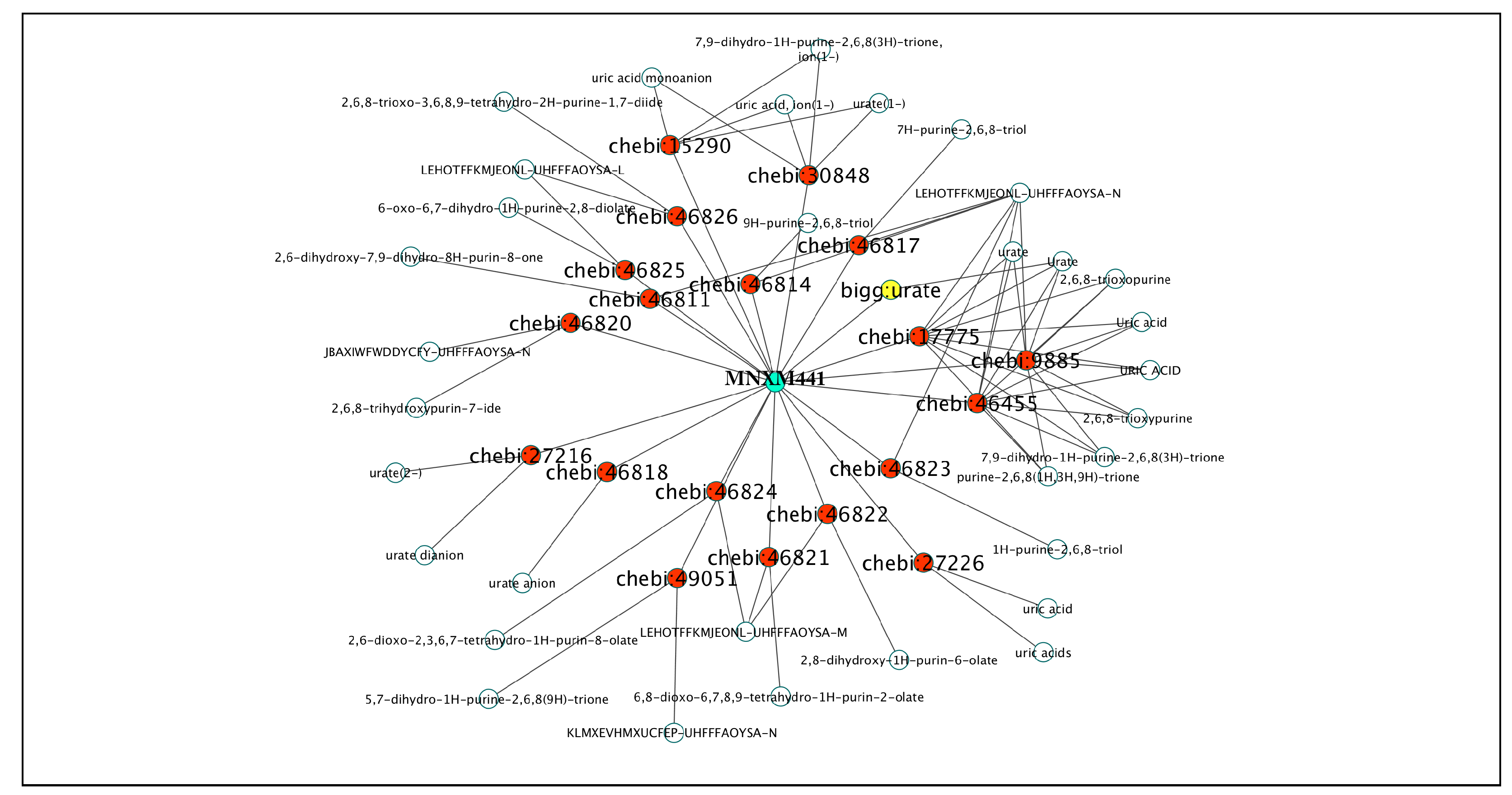
- Ambiguous. The Merriam-Webster dictionary defines ambiguous (second entry) as ‘capable of being understood in two or more possible senses or ways’. Here, we use ambiguous (and its derivatives) to refer to the case on which the same name links to more than one ID in the same database. An example is shown in Figure 1B, where the name ‘H’ links to the MetaCyc IDs ‘PROTON’ and ‘HIS’, associated with ‘hydrogen ion’ and ‘L-histidine’, respectively.
- Consistency: We use consistency (and its derivatives such as consistent) to refer to mappings on which a molecular entity is mapped to itself. It follows that inconsistency is used to indicate a mapping or a database on which a molecular entity is associated with a different one.
2.1. Mappings within the Same Database
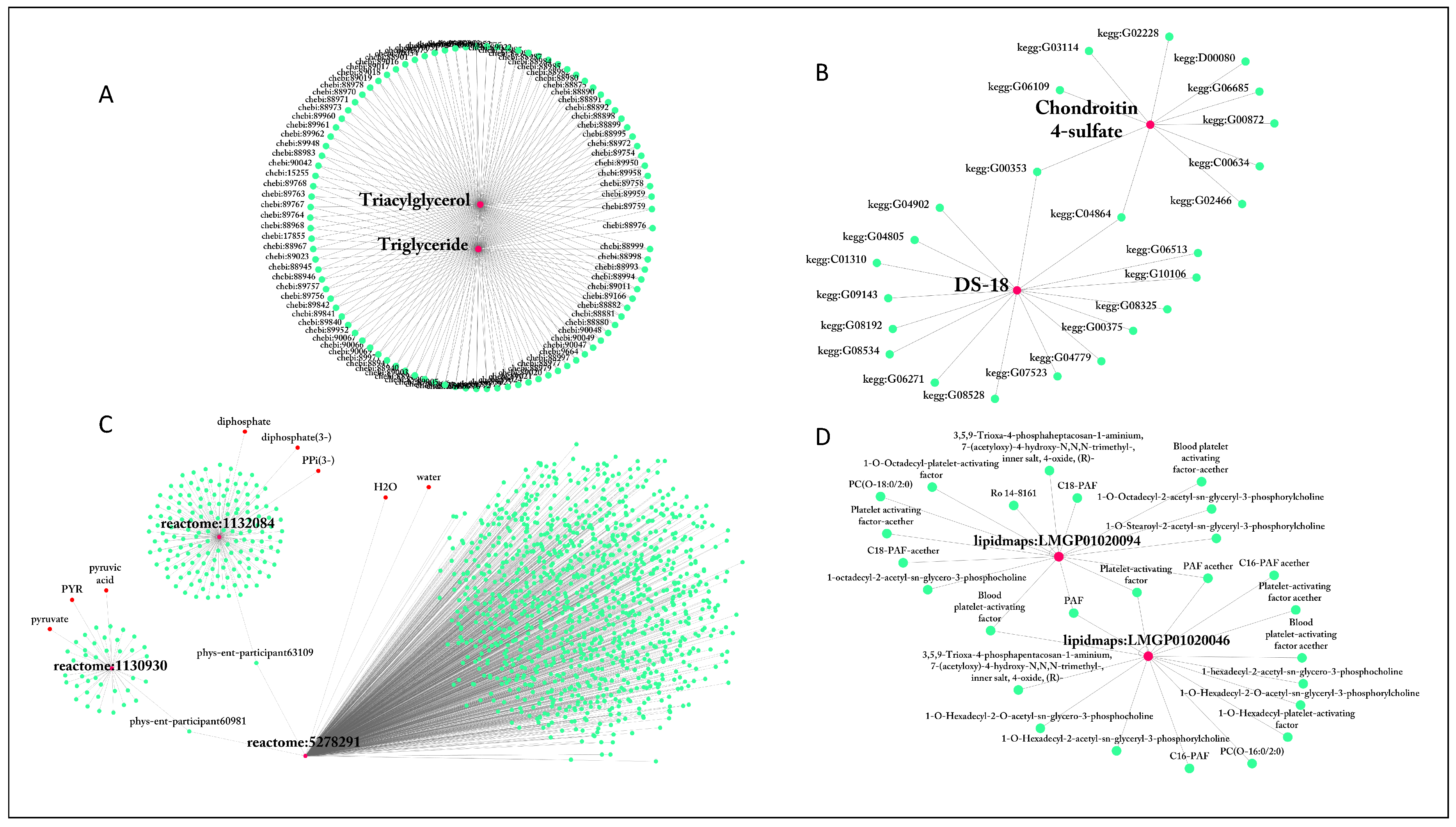
2.1.1. Name Ambiguity
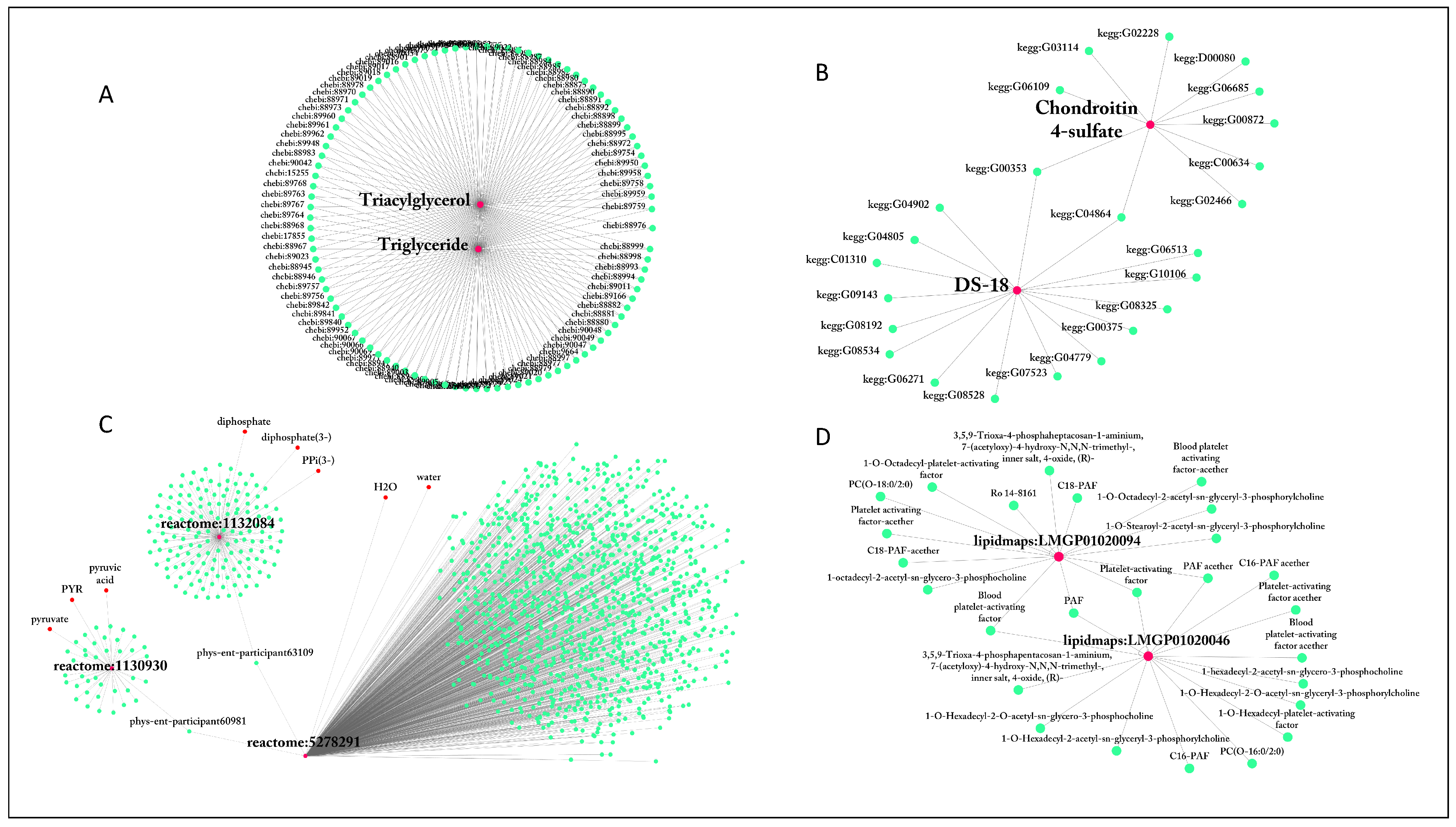
2.1.2. ID Multiplicity and Use of Synonyms
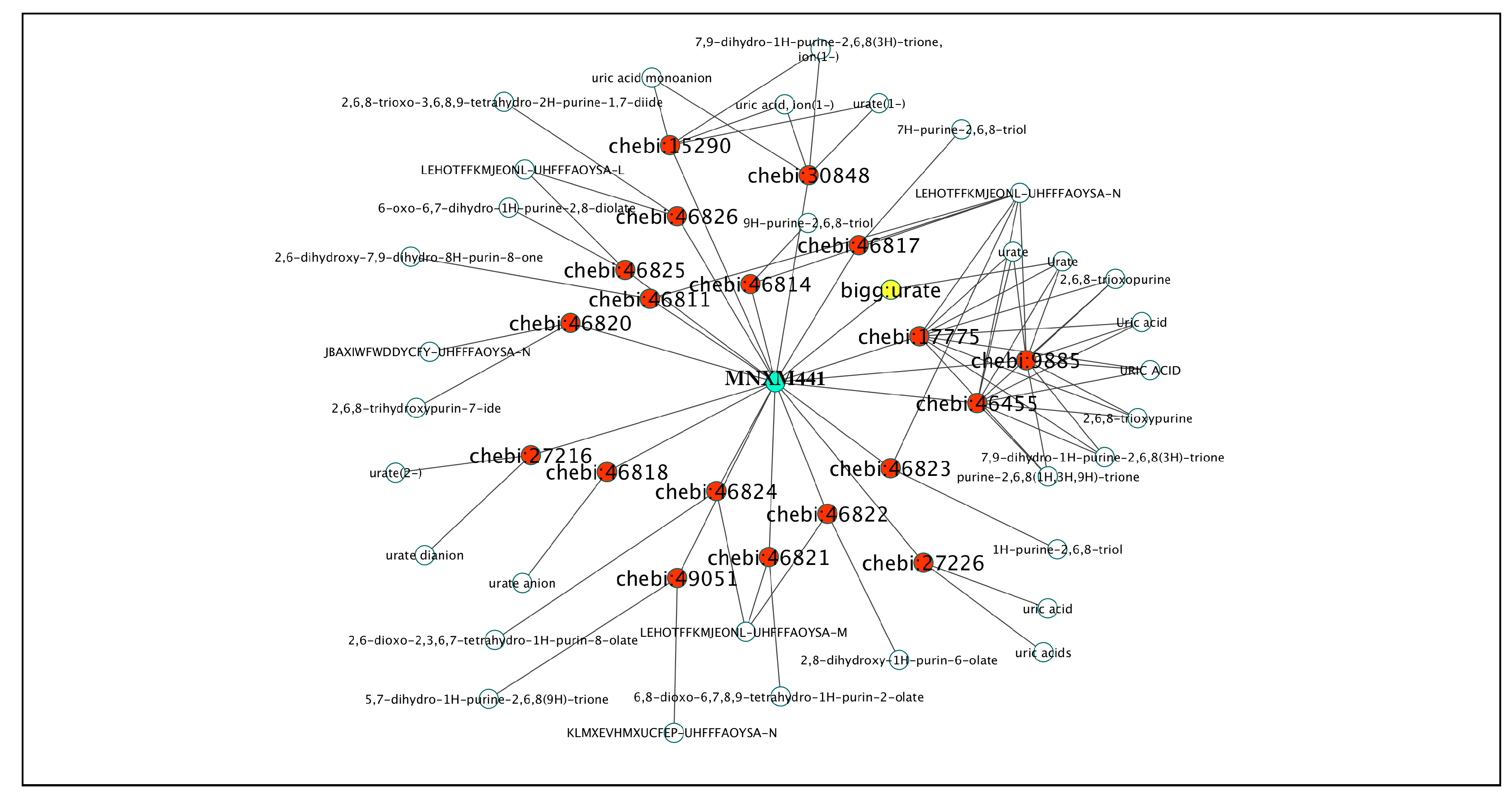
2.1.3. Database Mapping to IDs from MNXRef
2.2. Namespace Mapping between Databases
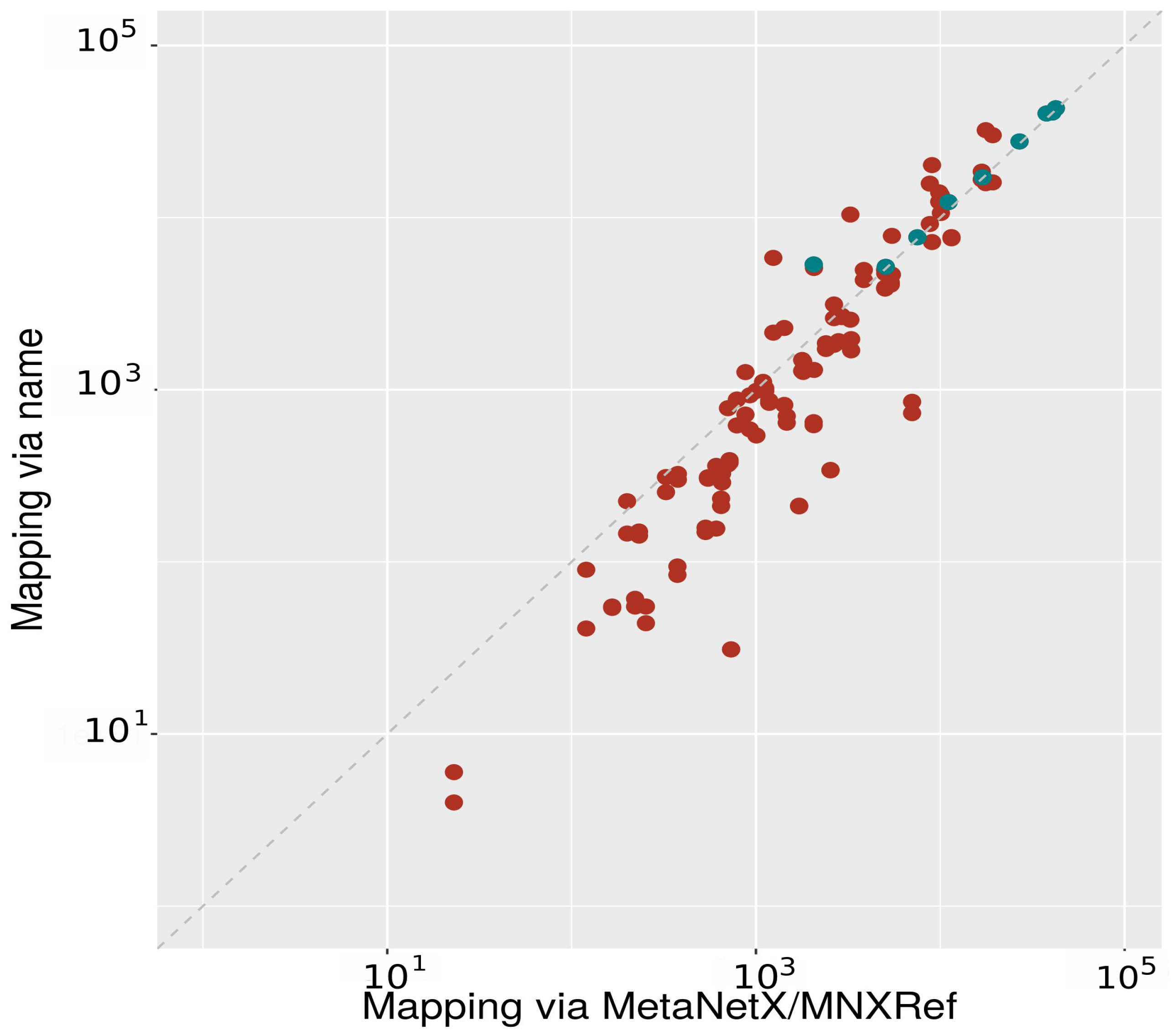
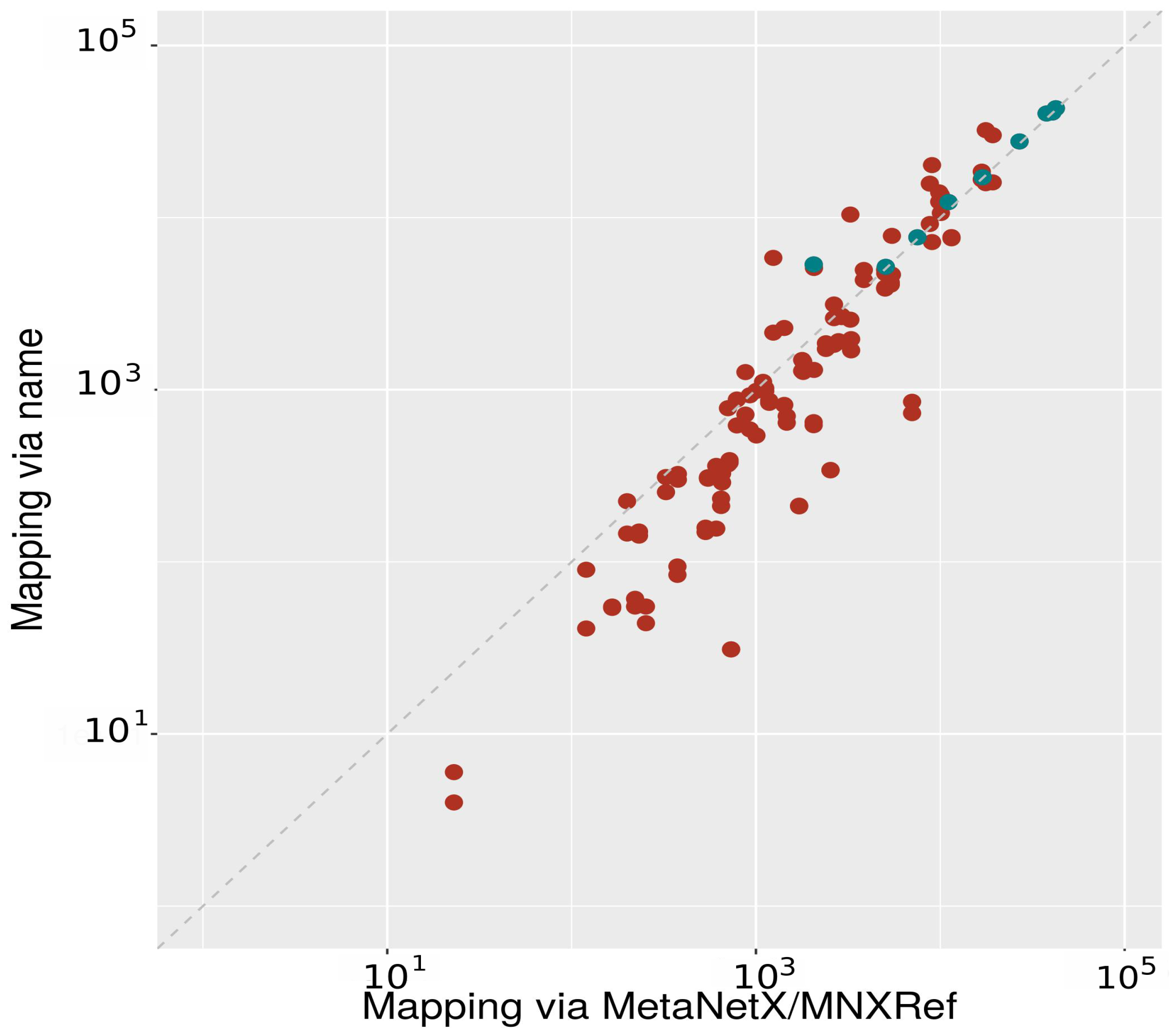
2.2.1. Mapping between Databases Using Metabolite Names
2.2.2. Mapping between Databases Using MNXRef ID
3. Discussion
- Limit the use of aliases, i.e., compound classes or abbreviations, as synonyms in databases. These aliases increase human readability, but should be clearly distinguished from names and synonyms in the databases and should not be used for mapping.
- In the context of metabolic modelling it is frequent and desirable to use compound classes to identify generic compounds [28]. Compounds such as ‘biomass’ or ‘lipid’ are often used in GEMs; this does not affect the use of the model, except when predicting or simulating the production (of a specific component) of generic compounds, i.e., when ‘lipids’ are the main focus of the model. In fact, it is often better to use generic compounds whenever a specific compound is not needed, as they can be universal. For instance, ‘biomass’ has been used as a standard among the modelling community as an artificial compound that represents the growth objective of the cell [6,17]. Another reason is that often the precise identity of the compounds is not needed and there is a lack of experimental data for their characterization. Therefore, when using generic compounds, it is desirable to add extensive annotation to the model to clearly state which compounds they represent, and for which purpose they are used in the model. These generic compounds are among the most ambiguous entities in the 11 analyzed databases and we therefore advise to exclude them from any automatic mapping process.
- Avoid using highly ambiguous names as the sole description of the compound in the model. When referring to these compounds, clear annotation needs to be included to prevent mismatches and inconsistencies.
- In addition to human-readable identifiers and database-dependent identifiers, include database-identifiers, such as InChI [15,16], whenever possible for compounds with defined structures. Using InChI can help to fully automate the mapping [28]. However, it should be taken into account that mismatches and errors can also happen because identifiers can also link to incorrect InChI as shown in [29,35,38].
- Model mapping only based on metabolite information can imply certain mismatch due to differences in namespaces, even if systematic identifiers were used. Hence, different mapping strategies, i.e., mapping through encoding genes and network topology [19], should be used to complement name or identifier-based mapping.
- GEMs also need to have a unique standard annotation so that they generate the same output even when different tools are used for the simulation. Neal et al. [39] suggest that semantic annotation can help to store and combine models, but these models need to stick to a unique standard annotation format.
4. Material and Methods
4.1. Data Collection and Preprocessing
4.2. Intra-Database Analysis
4.3. Inter-Database Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GEM | Genome-scale metabolic model |
| ID | Identifier |
References
- Oberhardt, M.A.; Palsson, B.O.; Papin, J.A. Applications of genome-scale metabolic reconstructions. Mol. Syst. Biol. 2009, 5, 320. [Google Scholar] [CrossRef]
- Patil, K.R.; Åkesson, M.; Nielsen, J. Use of genome-scale microbial models for metabolic engineering. Curr. Opin. Biotechnol. 2004, 15, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hua, Q. Applications of Genome-Scale Metabolic Models in Biotechnology and Systems Medicine. Front. Physiol. 2015, 6, 413. [Google Scholar] [CrossRef]
- Contreras, A.; Ribbeck, M.; Gutiérrez, G.D.; Cañon, P.M.; Mendoza, S.N.; Agosin, E. Mapping the physiological response of Oenococcus oeni to ethanol stress using an extended genome-scale metabolic model. Front. Microbiol. 2018, 9, 291. [Google Scholar] [CrossRef]
- Gudmundsson, S.; Agudo, L.; Nogales, J. Applications of genome-scale metabolic models of microalgae and cyanobacteria in biotechnology. In Microalgae-Based Biofuels and Bioproducts; Elsevier: Amsterdam, The Netherlands, 2018; pp. 93–111. [Google Scholar]
- Thiele, I.; Palsson, B.Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, D.A.; Edirisinghe, J.; Henry, C.S.; Overbeek, R.; O’Connell, T.G.; Edwards, R.A. From DNA to FBA: How To build your own genome-scale metabolic model. Front. Microbiol. 2016, 7, 907. [Google Scholar] [CrossRef] [PubMed]
- DeJongh, M.; Formsma, K.; Boillot, P.; Gould, J.; Rycenga, M.; Best, A. Toward the automated generation of genome-scale metabolic networks in the SEED. BMC Bioinform. 2007, 8, 139. [Google Scholar] [CrossRef]
- Karp, P.D.; Paley, S.; Romero, P. The pathway tools software. Bioinformatics 2002, 18, S225–S232. [Google Scholar] [CrossRef]
- Agren, R.; Liu, L.; Shoaie, S.; Vongsangnak, W.; Nookaew, I.; Nielsen, J. The RAVEN toolbox and its use for generating a genome-scale metabolic model for Penicillium chrysogenum. PLoS Comput. Biol. 2013, 9, e1002980. [Google Scholar] [CrossRef]
- Faria, J.P.; Rocha, M.; Rocha, I.; Henry, C.S. Methods for automated genome-scale metabolic model reconstruction. Biochem. Soc. Trans. 2018, 46, 931–936. [Google Scholar] [CrossRef]
- Karp, P.D.; Riley, M.; Paley, S.M.; Pellegrini-Toole, A. The metacyc database. Nucleic Acids Res. 2002, 30, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. The KEGG database. In ‘In Silico’Simulation of Biological Processes: Novartis Foundation Symposium 247; Wiley Online Library: Hoboken, NJ, USA, 2002; Volume 247, pp. 91–103. [Google Scholar]
- Ravikrishnan, A.; Raman, K. Critical assessment of genome-scale metabolic networks: the need for a unified standard. Brief. Bioinform. 2015, 16, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Heller, S.; McNaught, A.; Stein, S.; Tchekhovskoi, D.; Pletnev, I. InChI-the worldwide chemical structure identifier standard. J. Cheminform. 2013, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Heller, S.R.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC international chemical identifier. J. Cheminform. 2015, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Lieven, C.; Beber, M.E.; Olivier, B.G.; Bergmann, F.T.; Babaei, P.; Bartell, J.A.; Blank, L.M.; Chauhan, S.; Correia, K.; Diener, C.; et al. Memote: A community-driven effort towards a standardized genome-scale metabolic model test suite. bioRxiv 2018, 350991. [Google Scholar] [CrossRef]
- Herrgård, M.J.; Swainston, N.; Dobson, P.; Dunn, W.B.; Arga, K.Y.; Arvas, M.; Blüthgen, N.; Borger, S.; Costenoble, R.; Heinemann, M.; et al. A consensus yeast metabolic network reconstruction obtained from a community approach to systems biology. Nat. Biotechnol. 2008, 26, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- van Heck, R.G.; Ganter, M.; dos Santos, V.A.M.; Stelling, J. Efficient reconstruction of predictive consensus metabolic network models. PLoS Comput. Biol. 2016, 12, e1005085. [Google Scholar] [CrossRef]
- Reed, J.L. Genome-scale metabolic modeling and its application to microbial communities. In The Chemistry of Microbiomes: Proceedings of a Seminar Series; National Academies Press: Washington, DC, USA, 2017. [Google Scholar]
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018, 46, 7542–7553. [Google Scholar] [CrossRef]
- Mednis, M.; Vigants, A. Automatic comparison of metabolites names: Impact of criteria thresholds. Biosyst. Inf. Technol. 2013, 2, 1–5. [Google Scholar] [CrossRef]
- Qi, X.; Ozsoyoglu, Z.M.; Ozsoyoglu, G. Matching metabolites and reactions in different metabolic networks. Methods 2014, 69, 282–297. [Google Scholar] [CrossRef]
- Moretti, S.; Martin, O.; Van Du Tran, T.; Bridge, A.; Morgat, A.; Pagni, M. MetaNetX/MNXref–reconciliation of metabolites and biochemical reactions to bring together genome-scale metabolic networks. Nucleic Acids Res. 2015, 44, D523–D526. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Suthers, P.F.; Maranas, C.D. MetRxn: A knowledgebase of metabolites and reactions spanning metabolic models and databases. BMC Bioinform. 2012, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Bernard, T.; Bridge, A.; Morgat, A.; Moretti, S.; Xenarios, I.; Pagni, M. Reconciliation of metabolites and biochemical reactions for metabolic networks. Brief. Bioinform. 2012, 15, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Haraldsdóttir, H.S.; Thiele, I.; Fleming, R.M. Comparative evaluation of open source software for mapping between metabolite identifiers in metabolic network reconstructions: Application to Recon 2. J. Cheminform. 2014, 6, 2. [Google Scholar] [CrossRef]
- Williams, A.J.; Ekins, S.; Tkachenko, V. Towards a gold standard: Regarding quality in public domain chemistry databases and approaches to improving the situation. Drug Discov. Today 2012, 17, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Redestig, H.; Kusano, M.; Fukushima, A.; Matsuda, F.; Saito, K.; Arita, M. Consolidating metabolite identifiers to enable contextual and multi-platform metabolomics data analysis. BMC Bioinform. 2010, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Akhondi, S.A.; Muresan, S.; Williams, A.J.; Kors, J.A. Ambiguity of non-systematic chemical identifiers within and between small-molecule databases. J. Cheminform. 2015, 7, 54. [Google Scholar] [CrossRef]
- Labena, A.A.; Gao, Y.Z.; Dong, C.; Hua, H.L.; Guo, F.B. Metabolic pathway databases and model repositories. Quant. Biol. 2018, 6, 30–39. [Google Scholar] [CrossRef]
- Latendresse, M. Efficiently gap-filling reaction networks. BMC Bioinform. 2014, 15, 225. [Google Scholar] [CrossRef]
- Christian, N.; May, P.; Kempa, S.; Handorf, T.; Ebenhöh, O. An integrative approach towards completing genome-scale metabolic networks. Mol. BioSyst. 2009, 5, 1889–1903. [Google Scholar] [CrossRef] [PubMed]
- Akhondi, S.A.; Kors, J.A.; Muresan, S. Consistency of systematic chemical identifiers within and between small-molecule databases. J. Cheminform. 2012, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Gottstein, W.; Olivier, B.G.; Bruggeman, F.J.; Teusink, B. Constraint-based stoichiometric modelling from single organisms to microbial communities. J. R. Soc. Interface 2016, 13, 20160627. [Google Scholar] [CrossRef] [PubMed]
- van der Ark, K.C.H.; van Heck, R.G.A.; Dos Santos, V.A.P.M.; Belzer, C.; de Vos, W.M. More than just a gut feeling: constraint-based genome-scale metabolic models for predicting functions of human intestinal microbes. Microbiome 2017, 5, 78. [Google Scholar] [CrossRef] [PubMed]
- Young, D.; Martin, T.; Venkatapathy, R.; Harten, P. Are the chemical structures in your QSAR correct? QSAR Comb. Sci. 2008, 27, 1337–1345. [Google Scholar] [CrossRef]
- Neal, M.L.; König, M.; Nickerson, D.; Mısırlı, G.; Kalbasi, R.; Dräger, A.; Atalag, K.; Chelliah, V.; Cooling, M.; Cook, D.L.; et al. Harmonizing semantic annotations for computational models in biology. bioRxiv 2018, 246470. [Google Scholar] [CrossRef] [PubMed]
- King, Z.A.; Lu, J.; Dräger, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsson, B.O.; Lewis, N.E. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2015, 44, D515–D522. [Google Scholar] [CrossRef]
- Devoid, S.; Overbeek, R.; DeJongh, M.; Vonstein, V.; Best, A.A.; Henry, C. Automated genome annotation and metabolic model reconstruction in the SEED and Model SEED. In Systems Metabolic Engineering; Springer: Berlin, Germany, 2013; pp. 17–45. [Google Scholar]
- Degtyarenko, K.; De Matos, P.; Ennis, M.; Hastings, J.; Zbinden, M.; McNaught, A.; Alcántara, R.; Darsow, M.; Guedj, M.; Ashburner, M. ChEBI: A database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2007, 36, D344–D350. [Google Scholar] [CrossRef]
- Wicker, J.; Lorsbach, T.; Gütlein, M.; Schmid, E.; Latino, D.; Kramer, S.; Fenner, K. enviPath—The environmental contaminant biotransformation pathway resource. Nucleic Acids Res. 2015, 44, D502–D508. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The human metabolome database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; et al. Lmsd: Lipid maps structure database. Nucleic Acids Res. 2006, 35, D527–D532. [Google Scholar] [CrossRef] [PubMed]
- Joshi-Tope, G.; Gillespie, M.; Vastrik, I.; D’Eustachio, P.; Schmidt, E.; de Bono, B.; Jassal, B.; Gopinath, G.; Wu, G.; Matthews, L.; et al. Reactome: A knowledgebase of biological pathways. Nucleic Acids Res. 2005, 33, D428–D432. [Google Scholar] [CrossRef] [PubMed]
- Wittig, U.; Kania, R.; Golebiewski, M.; Rey, M.; Shi, L.; Jong, L.; Algaa, E.; Weidemann, A.; Sauer-Danzwith, H.; Mir, S.; et al. SABIO-RK—Database for biochemical reaction kinetics. Nucleic Acids Res. 2011, 40, D790–D796. [Google Scholar] [CrossRef] [PubMed]
- Aimo, L.; Liechti, R.; Hyka-Nouspikel, N.; Niknejad, A.; Gleizes, A.; Götz, L.; Kuznetsov, D.; David, F.P.; van der Goot, F.G.; Riezman, H.; et al. The SwissLipids knowledgebase for lipid biology. Bioinformatics 2015, 31, 2860–2866. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | #Name | Average Number of IDs per Name ± s.d. | % Ambiguous Names | # Ambiguous Names | Highest Number of IDs per Name |
|---|---|---|---|---|---|
| BiGG | 5102 | 1.0141 ± 0.126 | 1.31 | 67 | 3 |
| ChEBI | 388,505 | 1.3846 ± 1.52 | 14.8 | 57,497 | 413 |
| enviPath | 11,648 | 1.0804 ± 0.325 | 7.38 | 860 | 10 |
| HMDB | 101,101 | 1.0377 ± 3.865 | 1.67 | 1686 | 921 |
| KEGG | 59,682 | 1.1461 ± 0.422 | 13.3 | 7936 | 16 |
| LIPID MAPS | 77,457 | 1.0113 ± 0.33 | 0.62 | 478 | 63 |
| MetaCyc | 55,823 | 1.0058 ± 0.103 | 0.5 | 279 | 13 |
| Reactome | 6972 | 1.7902 ± 2.458 | 29.43 | 2052 | 34 |
| SABIO-RK | 11,475 | 1.0008 ± 0.031 | 0.07 | 8 | 3 |
| SEED | 47,410 | 1.0108 ± 0.106 | 1.06 | 503 | 4 |
| SLM | 1,218,750 | 1.0782 ± 0.321 | 6.72 | 81,894 | 9 |
| Database | #ID | Average Multiplicity ± s.d. | % of IDs with Multiplicity > 1 | # of IDs with Multiplicity > 1 | Highest Multiplicity in Database |
|---|---|---|---|---|---|
| BiGG | 5174 | 1.0 ± 0.0 | 0.0 | 0 | 1 |
| ChEBI | 123,835 | 4.344 ± 3.588 | 97.74 | 121,034 | 57 |
| enviPath | 12,306 | 1.0226 ± 0.229 | 1.6 | 197 | 10 |
| HMDB | 43,179 | 2.4297 ± 0.512 | 99.71 | 43,052 | 8 |
| KEGG | 40,256 | 1.6991 ± 1.231 | 38.93 | 15,671 | 31 |
| LIPID MAPS | 40,772 | 3.9213 ± 0.962 | 100.0 | 40,772 | 23 |
| MetaCyc | 17,159 | 3.2722 ± 1.984 | 99.75 | 17,116 | 98 |
| Reactome | 5344 | 2.3355 ± 16.65 | 47.46 | 2536 | 1106 |
| SABIO-RK | 7683 | 1.4947 ± 1.193 | 24.17 | 1857 | 21 |
| SEED | 27,693 | 1.7305 ± 1.311 | 39.83 | 11,031 | 28 |
| SLM | 505,004 | 2.602 ± 0.611 | 99.87 | 504,333 | 9 |
| Metabolite Name | # Associate IDs | Metabolite ID | # Associated Names |
|---|---|---|---|
| lecithin | 922 | reactome:5278291 | 1106 |
| diacylglycerol | 812 | reactome:1131511 | 266 |
| Lecithin | 417 | reactome:1236709 | 266 |
| Diglyceride | 317 | reactome:1132345 | 180 |
| Diacylglycerol | 317 | reactome:1132084 | 155 |
| Triacylglycerol | 106 | reactome:1132304 | 140 |
| Triglyceride | 103 | reactome:5278409 | 123 |
| PPP | 66 | reactome:5278317 | 107 |
| Cer[NS] | 63 | MetaCyc:PARATHION | 98 |
| Database | #ID | #MNXRef ID | Average #ID per MNXRef ID ± s.d. | % of IDs with Multiplicity >1 | # of IDs with Multiplicity > 1 | Highest ID Multiplicity > 1 |
|---|---|---|---|---|---|---|
| BiGG | 5174 | 5062 | 1.0221 ± 0.165 | 1.96 | 99 | 4 |
| ChEBI | 123,835 | 96,746 | 1.28 ± 1.005 | 11.93 | 11,541 | 30 |
| enviPath | 12,306 | 11,087 | 1.1099 ± 0.44 | 8.14 | 902 | 9 |
| HMDB | 43,179 | 42,354 | 1.0195 ± 0.176 | 1.63 | 691 | 12 |
| KEGG | 40,256 | 37,722 | 1.0672 ± 0.293 | 6.14 | 2316 | 12 |
| LIPID MAPS | 40,772 | 40,546 | 1.0056 ± 0.083 | 0.51 | 207 | 6 |
| MetaCyc | 17,159 | 16,985 | 1.0102 ± 0.115 | 0.9 | 153 | 5 |
| Reactome | 5344 | 2058 | 2.5967 ± 3.895 | 41.93 | 863 | 34 |
| SABIO-RK | 7683 | 7512 | 1.0228 ± 0.154 | 2.2 | 165 | 3 |
| SEED | 27,693 | 26,894 | 1.0297 ± 0.181 | 2.79 | 749 | 4 |
| SLM | 505,004 | 504,881 | 1.0002 ± 0.016 | 0.02 | 119 | 3 |
| Database | BiGG | ChEBI | enviPath | HMDB | KEGG | LIPID MAPS | MetaCyc | Reactome | SABIO-RK | SEED | SLM |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BiGG | – | 5097 (4.1%) | 150 (1.2%) | 702 (1.6%) | 1489 (3.7%) | 158 (0.4%) | 210 (1.2%) | 361 (6.8%) | 839 (10.9%) | 1829 (6.6%) | 61 (0.0%) |
| ChEBI | 1303 (25.2%) | – | 816 (6.6%) | 9178 (21.3%) | 16013 (39.8%) | 4662 (11.4%) | 7209 (42.0%) | 2146 (40.2%) | 2552 (33.2%) | 15,837 (57.2%) | 4336 (0.9%) |
| enviPath | 142 (2.7%) | 2284 (1.8%) | – | 304 (0.7%) | 1111 (2.8%) | 55 (0.1%) | 31 (0.2%) | 90 (1.7%) | 300 (3.9%) | 983 (3.5%) | 6 (0.0%) |
| HMDB | 643 (12.4%) | 15,749 (12.7%) | 310 (2.5%) | – | 4745 (11.8%) | 4078 (10.0%) | 1693 (9.9%) | 877 (16.4%) | 1268 (16.5%) | 3868 (14.0%) | 14,007 (2.8%) |
| KEGG | 1286 (24.9%) | 30,098 (24.3%) | 1050 (8.5%) | 3922 (9.1%) | – | 1725 (4.2%) | 731 (4.3%) | 928 (17.4%) | 2604 (33.9%) | 16,646 (60.1%) | 84 (0.0%) |
| LIPID MAPS | 149 (2.9%) | 7832 (6.3%) | 54 (0.4%) | 4200 (9.7%) | 1862 (4.6%) | – | 622 (3.6%) | 311 (5.8%) | 377 (4.9%) | 1893 (6.8%) | 13,478 (2.7%) |
| MetaCyc | 212 (4.1%) | 20,183 (16.3%) | 31 (0.3%) | 1967 (4.6%) | 851 (2.1%) | 648 (1.6%) | – | 1266 (23.7%) | 340 (4.4%) | 7703 (27.8%) | 326 (0.1%) |
| Reactome | 156 (3.0%) | 5833 (4.7%) | 41 (0.3%) | 620 (1.4%) | 588 (1.5%) | 254 (0.6%) | 717 (4.2%) | – | 368 (4.8%) | 542 (2.0%) | 146 (0.0%) |
| SABIO-RK | 864 (16.7%) | 10,413 (8.4%) | 324 (2.6%) | 1456 (3.4%) | 3127 (7.8%) | 390 (1.0%) | 342 (2.0%) | 781 (14.6%) | – | 2692 (9.7%) | 55 (0.0%) |
| SEED | 1824 (35.3%) | 32,212 (26.0%) | 1020 (8.3%) | 4971 (11.5%) | 18,489 (45.9%) | 1915 (4.7%) | 7580 (44.2%) | 985 (18.4%) | 2641 (34.4%) | – | 233 (0.0%) |
| SLM | 55 (1.1%) | 4964 (4.0%) | 4 (0.0%) | 12,354 (28.6%) | 94 (0.2%) | 10,634 (26.1%) | 289 (1.7%) | 225 (4.2%) | 44 (0.6%) | 211 (0.8%) | – |
| Database | BiGG | ChEBI | enviPath | HMDB | KEGG | LIPID MAPS | MetaCyc | Reactome | SABIO-RK | SEED | SLM |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BiGG | – | 2.9 | 1.3 | 3.0 | 3.6 | 3.2 | 1.4 | 0.6 | 2.9 | 2.7 | 1.6 |
| ChEBI | 76.3 | – | 67.0 | 38.1 | 38.3 | 34.3 | 58.7 | 81.3 | 78.7 | 37.3 | 26.9 |
| enviPath | 6.3 | 6.5 | – | 8.2 | 6.1 | 0.0 | 0.0 | 12.2 | 7.7 | 4.6 | 0.0 |
| HMDB | 10.7 | 11.5 | 6.8 | – | 7.3 | 4.3 | 13.2 | 22.8 | 12.8 | 7.4 | 0.7 |
| KEGG | 17.0 | 15.2 | 11.1 | 28.5 | – | 10.2 | 18.5 | 34.5 | 19.6 | 12.4 | 33.3 |
| LIPID MAPS | 8.7 | 9.8 | 1.9 | 1.8 | 3.2 | – | 4.2 | 13.2 | 4.5 | 3.2 | 0.8 |
| MetaCyc | 0.5 | 3.9 | 0.0 | 2.5 | 3.9 | 2.0 | – | 6.0 | 4.1 | 1.5 | 0.6 |
| Reactome | 42.3 | 41.4 | 51.2 | 49.0 | 51.4 | 24.4 | 38.9 | – | 49.5 | 43.2 | 47.9 |
| SABIO-RK | 0.0 | 4.5 | 0.0 | 0.0 | 3.8 | 1.0 | 3.8 | 2.2 | – | 3.3 | 1.8 |
| SEED | 3.0 | 6.0 | 0.9 | 2.0 | 2.4 | 2.2 | 3.1 | 8.9 | 5.3 | – | 1.7 |
| SLM | 7.3 | 37.2 | 25.0 | 12.3 | 18.1 | 22.3 | 10.4 | 24.4 | 20.5 | 9.5 | – |
| Database | BiGG | ChEBI | enviPath | HMDB | KEGG | LIPID MAPS | MetaCyc | Reactome | SABIO-RK | SEED | SLM |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BiGG | – | 2064 (2.1%) | 232 (2.1%) | 1469 (3.5%) | 1781 (4.7%) | 533 (1.3%) | 1715 (10.1%) | 609 (29.6%) | 1180 (15.7%) | 2652 (9.9%) | 221 (0.0%) |
| ChEBI | 2064 (40.8%) | – | 1424 (12.8%) | 8775 (20.7%) | 19,244 (51.0%) | 5464 (13.5%) | 9019 (53.1%) | 1242 (60.3%) | 3252 (43.3%) | 17,649 (65.6%) | 3848 (0.8%) |
| enviPath | 232 (4.6%) | 1424 (1.5%) | – | 549 (1.3%) | 1093 (2.9%) | 166 (0.4%) | 733 (4.3%) | 120 (5.8%) | 377 (5.0%) | 1123 (4.2%) | 23 (0.0%) |
| HMDB | 1469 (29.0%) | 8775 (9.1%) | 549 (5.0%) | – | 5028 (13.3%) | 5387 (13.3%) | 3283 (19.3%) | 788 (38.3%) | 1804 (24.0%) | 5021 (18.7%) | 9870 (2.0%) |
| KEGG | 1781 (35.2%) | 19,244 (19.9%) | 1093 (9.9%) | 5028 (11.9%) | – | 2397 (5.9%) | 7030 (41.4%) | 926 (45.0%) | 2651 (35.3%) | 16,791 (62.4%) | 375 (0.1%) |
| LIPID MAPS | 533 (10.5%) | 5464 (5.6%) | 166 (1.5%) | 5387 (12.7%) | 2397 (6.4%) | – | 2056 (12.1%) | 325 (15.8%) | 719 (9.6%) | 2807 (10.4%) | 10,076 (2.0%) |
| MetaCyc | 1715 (33.9%) | 9019 (9.3%) | 733 (6.6%) | 3283 (7.8%) | 7030 (18.6%) | 2056 (5.1%) | – | 877 (42.6%) | 2538 (33.8%) | 11,502 (42.8%) | 655 (0.1%) |
| Reactome | 609 (12.0%) | 1242 (1.3%) | 120 (1.1%) | 788 (1.9%) | 926 (2.5%) | 325 (0.8%) | 877 (5.2%) | – | 705 (9.4%) | 1006 (3.7%) | 200 (0.0%) |
| SABIO-RK | 1180 (23.3%) | 3252 (3.4%) | 377 (3.4%) | 1804 (4.3%) | 2651 (7.0%) | 719 (1.8%) | 2538 (14.9%) | 705 (34.3%) | – | 2915 (10.8%) | 253 (0.1%) |
| SEED | 2652 (52.4%) | 17,649 (18.2%) | 1123 (10.1%) | 5021 (11.9%) | 16,791 (44.5%) | 2807 (6.9%) | 11,502 (67.7%) | 1006 (48.9%) | 2915 (38.8%) | – | 647 (0.1%) |
| SLM | 221 (4.4%) | 3848 (4.0%) | 23 (0.2%) | 9870 (23.3%) | 375 (1.0%) | 10,076 (24.9%) | 655 (3.9%) | 200 (9.7%) | 253 (3.4%) | 647 (2.4%) | – |
| Database | BiGG | ChEBI | enviPath | HMDB | KEGG | LIPID MAPS | MetaCyc | Reactome | SABIO-RK | SEED | SLM |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BiGG | – | 3.9 | 5.2 | 3.5 | 3.9 | 3.2 | 4.0 | 3.8 | 4.5 | 3.2 | 2.7 |
| ChEBI | 83.1 | – | 56.2 | 39.7 | 36.4 | 37.8 | 64.7 | 76.8 | 72.2 | 39.4 | 27.8 |
| enviPath | 9.9 | 10.6 | – | 12.0 | 8.1 | 8.4 | 7.6 | 14.2 | 11.1 | 8.1 | 8.7 |
| HMDB | 19.1 | 6.8 | 12.6 | – | 9.3 | 5.1 | 12.7 | 26.4 | 17.2 | 9.7 | 1.6 |
| KEGG | 15.0 | 10.0 | 11.0 | 22.1 | – | 8.4 | 9.6 | 19.7 | 17.5 | 11.2 | 14.7 |
| LIPID MAPS | 10.5 | 2.8 | 6.0 | 2.5 | 4.5 | – | 4.6 | 14.5 | 7.0 | 4.2 | 0.5 |
| MetaCyc | 3.6 | 1.4 | 3.4 | 2.1 | 1.7 | 0.9 | – | 4.3 | 2.8 | 1.1 | 0.5 |
| Reactome | 42.7 | 33.3 | 45.0 | 32.4 | 37.1 | 28.9 | 36.7 | – | 41.7 | 37.2 | 35.0 |
| SABIO-RK | 8.1 | 4.7 | 5.6 | 6.1 | 5.3 | 3.8 | 5.6 | 9.2 | – | 5.1 | 4.7 |
| SEED | 8.4 | 3.5 | 4.2 | 4.6 | 3.7 | 3.6 | 5.7 | 12.1 | 9.5 | – | 5.1 |
| SLM | 5.0 | 1.1 | 0.0 | 0.2 | 5.6 | 0.4 | 2.7 | 6.0 | 5.1 | 3.2 | – |
| Abbreviation | Database | IDs in Database | MetaNetX ID | Compound(s) |
|---|---|---|---|---|
| suc | MetaCyc | SUC | MNXM25 | succinate |
| suc | Reactome | 188980 | MNXM167 | sucrose |
| H | MetaCyc | PROTON | MNXM1 | proton |
| H | MetaCyc | HIS | MNXM134 | L-histidine |
| tmp | BiGG | tmp | MNXM87343 | TMP |
| tmp | ChEBI | 10529 | MNXM257 | Thymidine monophosphate |
| tmp | KEGG | C01081 | MNXM662 | Thiamine monophosphate |
| tmp | MetaCyc | CPD-610 | MNXM88031 | cyclo-triphosphoric acid |
| PPP | Reactome | 1475054 | MNXM3109 | triphosphate ion |
| PPP | MetaCyc | 2-PHENYL-2-1-PIPERDINYLPROPANE | MNXM150634 | 2-phenyl-2-1piperdinylpropane |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, N.; van Heck, R.G.A.; van Dam, J.C.J.; Schaap, P.J.; Saccenti, E.; Suarez-Diez, M. Consistency, Inconsistency, and Ambiguity of Metabolite Names in Biochemical Databases Used for Genome-Scale Metabolic Modelling. Metabolites 2019, 9, 28. https://doi.org/10.3390/metabo9020028
Pham N, van Heck RGA, van Dam JCJ, Schaap PJ, Saccenti E, Suarez-Diez M. Consistency, Inconsistency, and Ambiguity of Metabolite Names in Biochemical Databases Used for Genome-Scale Metabolic Modelling. Metabolites. 2019; 9(2):28. https://doi.org/10.3390/metabo9020028
Chicago/Turabian StylePham, Nhung, Ruben G. A. van Heck, Jesse C. J. van Dam, Peter J. Schaap, Edoardo Saccenti, and Maria Suarez-Diez. 2019. "Consistency, Inconsistency, and Ambiguity of Metabolite Names in Biochemical Databases Used for Genome-Scale Metabolic Modelling" Metabolites 9, no. 2: 28. https://doi.org/10.3390/metabo9020028
APA StylePham, N., van Heck, R. G. A., van Dam, J. C. J., Schaap, P. J., Saccenti, E., & Suarez-Diez, M. (2019). Consistency, Inconsistency, and Ambiguity of Metabolite Names in Biochemical Databases Used for Genome-Scale Metabolic Modelling. Metabolites, 9(2), 28. https://doi.org/10.3390/metabo9020028