Genomic Survey, Transcriptome, and Metabolome Analysis of Apocynum venetum and Apocynum hendersonii to Reveal Major Flavonoid Biosynthesis Pathways

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Results and Discussion
2.1. Genome Survey of A. venetum and A. hendersonii
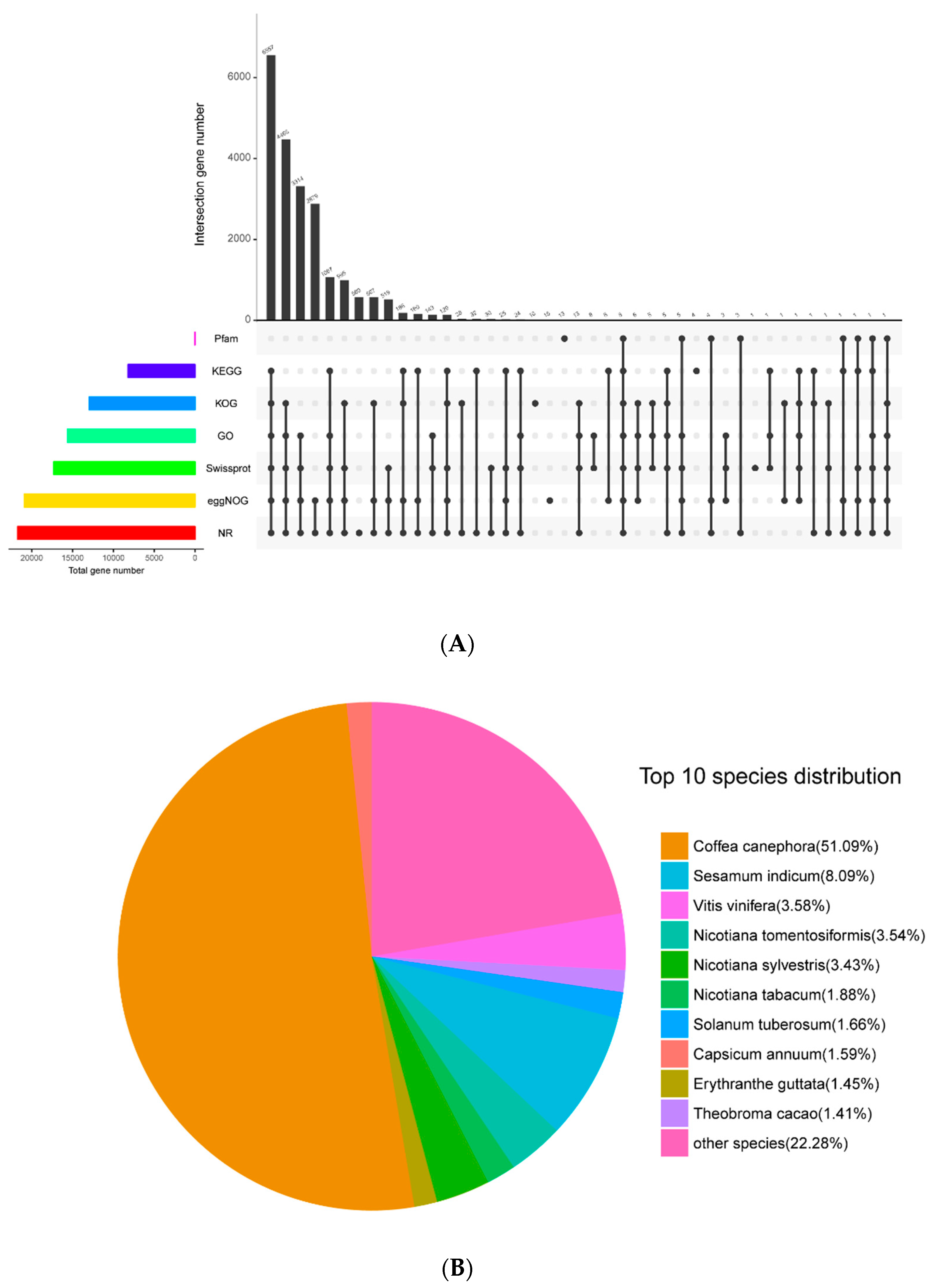
2.2. Transcriptome Sequencing, De Novo Assembly, and Pathway Analysis
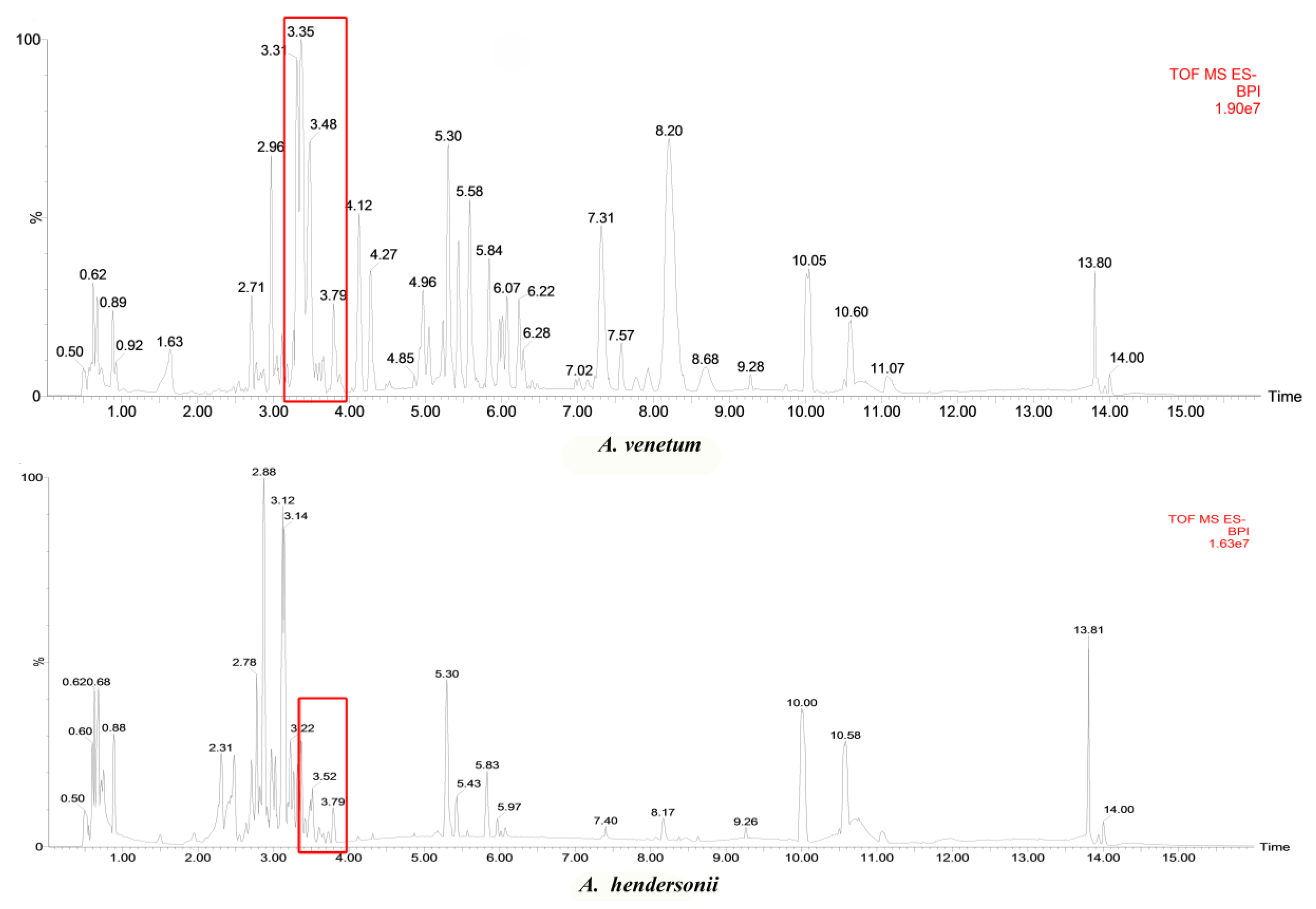
2.3. Metabolic Profiles of A. venetum and A. hendersonii
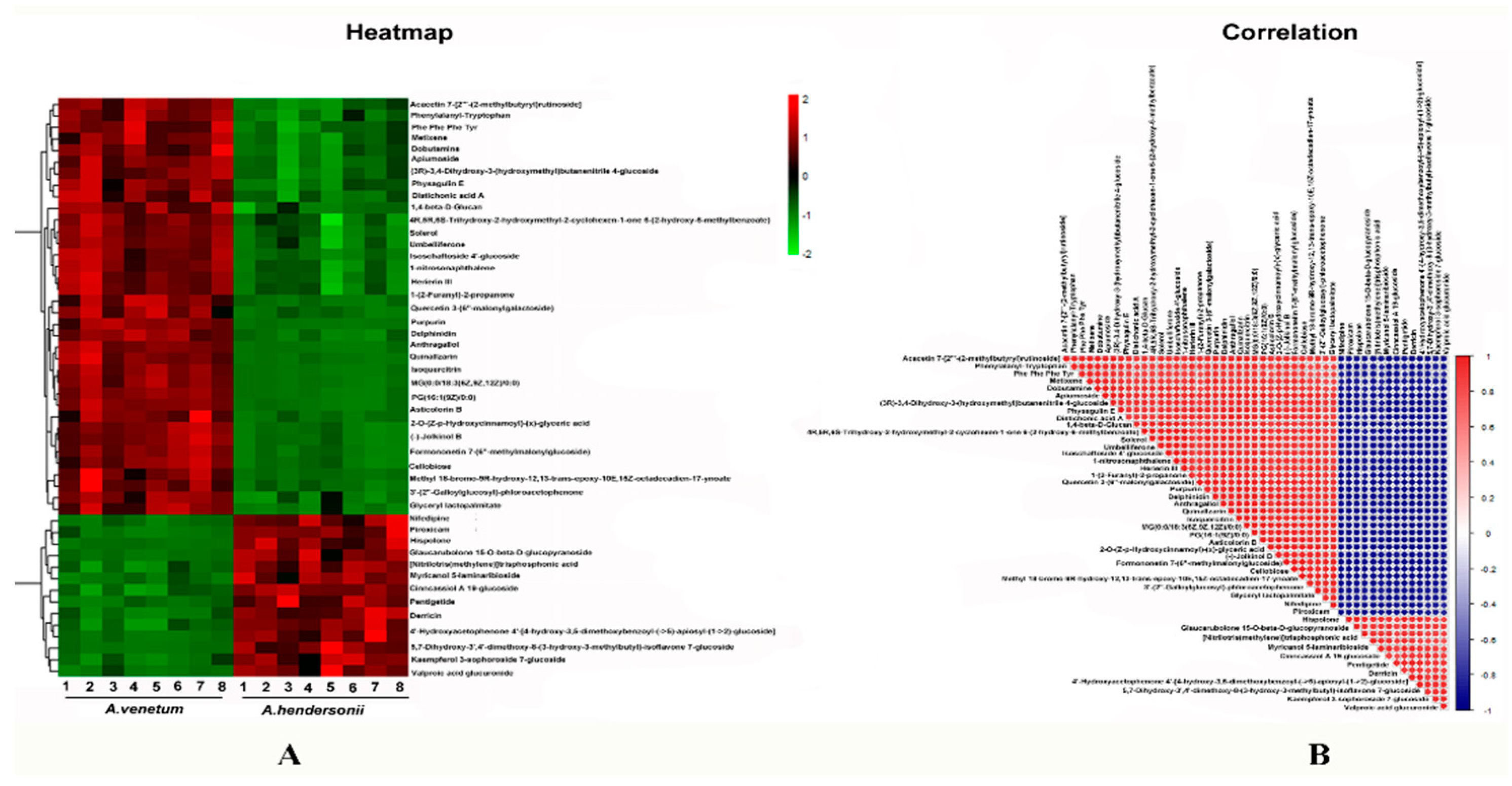
2.4. Major Flavonoids Identified in A. venetum and A. hendersonii
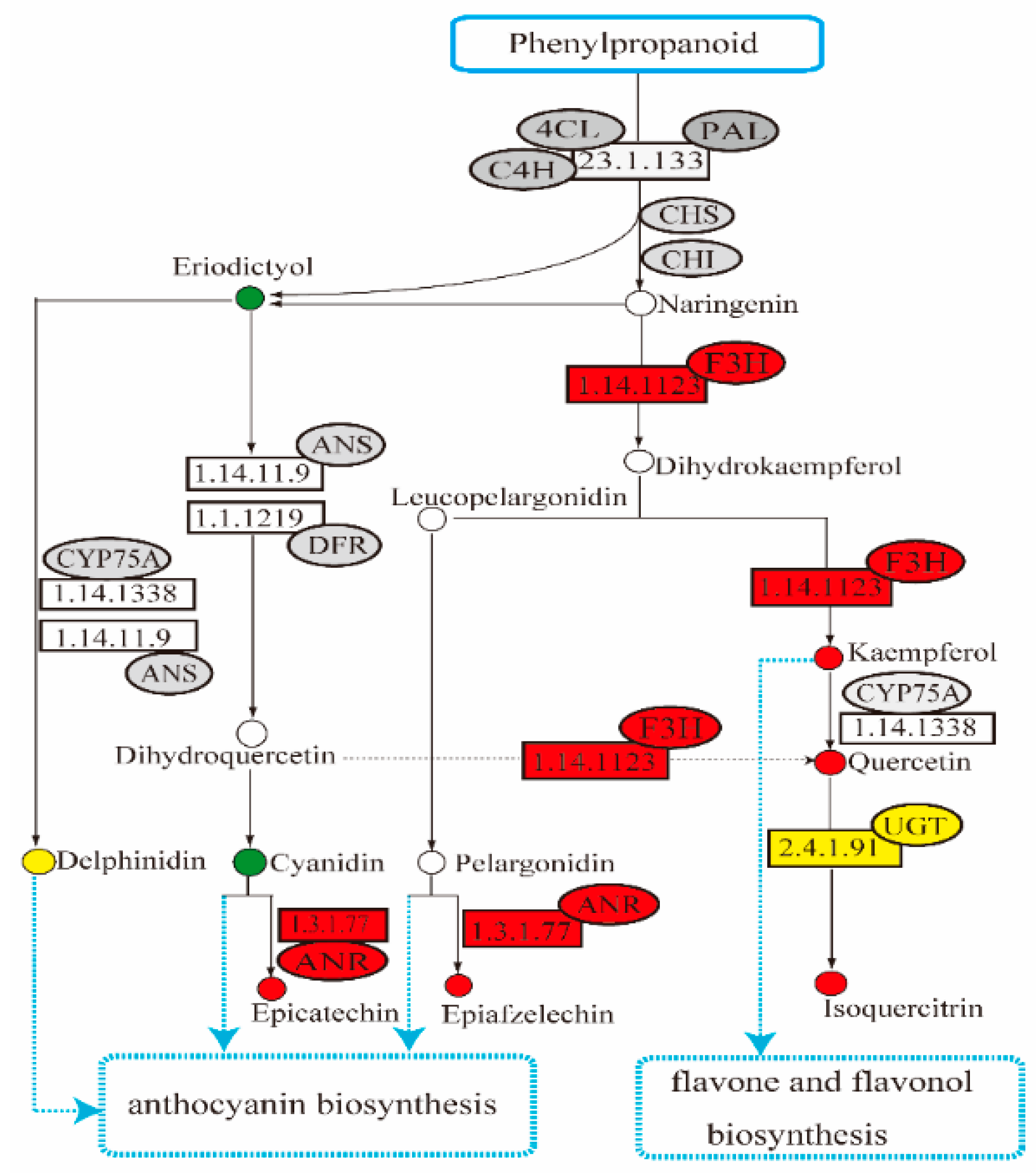
2.5. Analysis of Flavonoids Biosynthesis-Related Transcript Level and Metabolites Content
3. Materials and Methods
3.1. Plant Materials and Experimental Conditions
3.2. Genomic Survey, De Novo Transcriptome Assembly, and Analysis
3.3. Gene Function Annotation, Classification, and Digital Gene Expression Profile
3.4. LC/MS, Data Preprocessing, and Statistical Analysis
3.5. Flavonoids and Total Anthocyanin Quantifiation
3.6. Transcript–Metabolite Correlation Analysis and qRT-PCR Validation of the Selected Key Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Grundmann, O.; Nakajima, J.I.; Kamata, K.; Seo, S.; Butterweck, V. Kaempferol from the leaves of Apocynum venetum possesses anxiolytic activities in the elevated plus maze test in mice. Phytomedicine 2009, 16, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhang, W.; Peng, X.; Gu, G.; Chen, M.; Tang, Z. Development of Randomly Amplified Polymorphic DNA-Sequence Characterized Amplified Region Marker for Identification of Apocynum venetum LINN. from A. pictum SCHRENK. Biol. Pharm. Bull. 2010, 33, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Thevs, N.; Zerbe, S.; Kyosev, Y.; Rozi, A.; Tang, B.; Abdusalih, N.; Novitskij, Z. Apocynum venetum L. and Apocynum pictum Schrenk (Apocynaceae) as multi-functional and multi-service plant species in Central Asia: A review on biology, ecology, andutilization. J. Appl. Bot. Food Qual. 2012, 85, 159–167. [Google Scholar]
- Kasimu, R.; Fan, Z.; Wang, X.; Hu, J.; Wang, P.; Wang, J. Anti-platelet aggregation activities of different fractions in leaves of Apocynum venetum L. J. Ethnopharmacol. 2015, 168, 116–121. [Google Scholar] [CrossRef]
- Wu, T.; Li, X.; Li, T.; Cai, M.; Yu, Z.; Zhang, J.; Zhang, Z.; Zhang, W.; Xiang, J.; Cai, D. Apocynum venetum Leaf Extract Exerts Antidepressant-Like Effects and Inhibits Hippocampal and Cortical Apoptosis of Rats Exposed to Chronic Unpredictable Mild Stress. Evid. Based Complement. Altern. Med. 2018, 2, 1–8. [Google Scholar] [CrossRef]
- Lau, Y.S.; Kwan, C.Y.; Ku, T.C.; Hsieh, W.T.; Wang, H.D.; Nishibe, S.; Dharmani, M.; Mustafa, M.R. Apocynum venetum leaf extract, an antihypertensive herb, inhibits rat aortic contraction induced by angiotensin II: A nitric oxide and superoxide connection. J. Ethnopharmacol. 2012, 143, 565–571. [Google Scholar] [CrossRef]
- Chan, C.O.; Lau, C.C.; Ng, Y.F.; Xu, L.; Chen, S.B.; Chan, S.W.; Mok, K.W. Discrimination between Leave of Apocynum venetum and Its Adulterant, A. pictum Based on Antioxidant Assay and Chemical Profiles Combined with Multivariate Statistical Analysis. Antioxidants 2015, 4, 359–372. [Google Scholar] [CrossRef]
- Xie, W.; Zhang, X.Y.; Wang, T.; Hu, J. Botany, traditional uses, phytochemistry and pharmacology of Apocynum venetum L. (Luobuma): A review. J. Ethnopharmacol. 2012, 141, 1–8. [Google Scholar] [CrossRef]
- Yokozawa, T.; Kashiwada, Y.; Hattori, M.; Chung, H.Y. Study on the components of luobuma with peroxynitrite-scavenging activity. Biol. Pharm. Bull. 2012, 25, 748–752. [Google Scholar] [CrossRef]
- Zhang, W.O.; Peng, X.M.; Lu, C.M.; Wang, M.L.; Gu, G.P. A taxonomic study of luonuma based on sequence data of 3 non-coding DNA regions. Acta Bot. Boreali-Occident. Sin. 2007, 27, 859–863. [Google Scholar]
- Bogs, J.; Ebadi, A.; McDavid, D.; Robinson, S.P. Identification of the flavonoid hydroxylases from grapevine and their regulation during fruit development. Plant Physiol. 2006, 140, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Boss, P.K.; Davies, C.; Robinson, S.P. Analysis of the expression of anthocyanin pathway genes in developing Vitis vinifera L. cv shiraz grape berries and the implications for pathway regulation. Plant Physiol. 1996, 111, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Castellarin, S.D.; Pfeiffer, A.; Sivilotti, P.; Degan, M.; Peterlunger, E.; Gaspero, G.D. Transcriptional regulation of anthocyanin biosynthesis in ripening fruits of grapevine under seasonal water deficit. Plant Cell Environ. 2010, 30, 1381–1399. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.; Leitch, I.J.; Anadon-Rosell, A.; Canela, M.; Gálvez, F.; Garnatje, T.; Gras, A.; Hidalgo, O.; Bennett, M.D. Recent updates and developments to plant genome size databases. Nucleic Acids Res. 2014, 42, D1159–D1166. [Google Scholar] [CrossRef] [PubMed]
- Leitch, I.J.; Leitch, A.R. Genome Size Diversity and Evolution in Land Plants. Plant Genome Divers. 2013, 2, 307–322. [Google Scholar]
- Jiao, W.B.; Huang, D.; Xing, F.; Hu, Y.; Deng, X.X.; Xu, Q.; Chen, L.-L. Genome-wide characterization and expression analysis of genetic variants in sweet orange. Plant J. 2013, 75, 954–964. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, X.; Ke, C.; Du, H.; Ouyang, Y.; Chen, J.; Qiu, S.; Huang, J.; Jiang, Y.; Jiang, L.; et al. A killer-protector system regulates both hybrid sterility and segregation distortion in rice. Science 2012, 337, 1336–1340. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef]
- Belmonte, V.N.; Retamal, M.; Mezcua, M.; Fernández-Alba, A.R. A sensitive and selective method for the determination of selected pesticides in fruit by gas chromatography/mass spectrometry with negative chemical ionization. J. Chromatogr. A 2012, 1264, 110–116. [Google Scholar] [CrossRef]
- Liang, S.; Zhao, L.; Lv, L.; Zhang, H.; Guo, T.; Chai, Y.; Zhang, G. Screening and analysis of metabolites in rat urine after oral administration of Apocynum venetum L. extracts using HPLC–TOF-MS. J. Sep. Sci. 2014, 37, 515–526. [Google Scholar]
- Yan, S.X.; Lang, J.L.; Song, Y.Y.; Wu, Y.Z.; Lv, M.H.; Zhao, X.; Liu, Y.H.; Xu, C.Y. Studies on Anti-Depressant Activity of Four Flavonoids Isolated from Apocynum venetum Linn (Apocynaceae) Leaf in Mice. Trop. J. Pharm. Res. 2015, 14, 2269–2277. [Google Scholar] [CrossRef]
- Schaefer, H.; Schaefer, V.; Levey, D. How plant-animal interactions signal new insights in communication. Trends Ecol. Evol. 2004, 19, 0–584. [Google Scholar] [CrossRef]
- Bendokas, V.; Skemiene, K.; Trumbeckaite, S.; Stanys, V.; Passamonti, S.; Borutaite, V.; Liobikas, J. Anthocyanins: From plant pigments to health benefits at mitochondrial level. Crit. Rev. Food Sci. Nutr. 2019, 1, 1–14. [Google Scholar] [CrossRef]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Espley, R.V.; Hellens, R.P.; Putterill, J.; Stevenson, D.E.; Kutty-Amma, S.; Allan, A.C. Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 2010, 49, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, H.; Wang, N.; Jiang, S.; Liu, J.; Wang, D.; Zuo, W.; Chen, X. Molecular cloning and expression analysis of an auxin signaling related gene mdarf3 in red flesh apple. Acta Hortic. Sin. 2017, 44, 633–643. [Google Scholar]
- Wang, N.; Jiang, S.; Zhang, Z.; Fang, H.; Xu, H.; Wang, Y.; Chen, X. Malus sieversii: The origin, flavonoid synthesis mechanism, and breeding of red-skinned and red-fleshed apples. Hortic. Res. 2018, 5. [Google Scholar] [CrossRef]
- Su, M.; Wang, N.; Jiang, S.; Fang, H.; Xu, H.; Wang, Y.; Zhang, Z.; Zhang, J.; Xu, L.; Zhang, Z.; et al. Molecular characterization and expression analysis of the critical floral gene mdagl24-like in red-fleshed apple. Plant Sci. 2018, 276, 189–198. [Google Scholar] [CrossRef]
- Dong, T.; Han, R.; Yu, M.; Zhang, Y.; Gong, Y.; Li, Z. Anthocyanins accumulation and molecular analysis of correlated genes by metabolome and transcriptome in green and purple asparaguses (Asparagus officinalis, L.). Food Chem. 2018, 271, 18–28. [Google Scholar] [CrossRef]
- Huang, X.; Zou, X.; Zhao, J.; Shi, J.; Zhang, X.; Mel, H. Measurement of total anthocyanins content in flowering tea using near infrared spectroscopy combined with ant colony optimization models. Food Chem. 2014, 164, 536–543. [Google Scholar]
- Ye, J.; Zhang, Y.; Cui, H.; Liu, J.; Wu, Y.; Cheng, Y.; Xu, H.; Huang, X.; Li, S.; Zhou, A.; et al. Wego 2.0: A web tool for analyzing and plotting go annotations, 2018 update. Nucleic Acids Res. 2018, 2, W71–W75. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Shi, Y.; Yuan, J.; Hu, X.; Zhang, H.; Li, N.; Li, Z.; Chen, Y.; Mu, D.; Fan, W. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv 2013, arXiv:1308.2012. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Sagaradze, V.A.; Babaeva, E.Y.; Kalenikova, E.I. HPLC-UV method for determing flavonoids in hawthorn flowers and leaves. Pharm. Chem. J. 2017, 51, 277–280. [Google Scholar] [CrossRef]
- Patil, G.; Madhusudhan, M.C.; Babu, B.R.; Raghavarao, K. Extraction, dealcoholization and concentration of anthocyanin from red radish. Chem. Eng. Process. Process Intensif. 2009, 48, 364–369. [Google Scholar] [CrossRef]
- Tsantili, E.; Shin, Y.; Nock, J.F.; Watkins, C.B. Antioxidant concentrations during chilling injury development in peaches. Postharvest Biol. Technol. 2010, 57, 27–34. [Google Scholar] [CrossRef]
- White, B.L.; Howard, L.R.; Prior, R.L. Impact of Different Stages of Juice Processing on the Anthocyanin, Flavonol, and Procyanidin Contents of Cranberries. J. Agric. Food Chem. 2011, 59, 4692–4698. [Google Scholar] [CrossRef]
- Inácio, M.R.; De-Lima, K.M.; Michchell, G.; Lopes, V.G.; Pessoa, J.D.; Henrique, D.A. Total anthocyanin content determination in intact a ç aí (Euterpe oleracea Mart.) and palmitero-juara (Euterpe edulis Mart.) fruit using near infrared spectroscopy (NIR) and multivariate calibration. Food Chem. 2013, 136, 1160–1164. [Google Scholar] [CrossRef]
- Hur, M.; Campbell, A.A.; Almeida-de-Macedo, M.; Li, L.; Ransom, N.; Jose, A.; Crispin, M.; Nikolau, B.J.; Wurtele, E.S. A global approachto analysis and interpretation of metabolic data for plant natural product discovery. Nat. Prod. Rep. 2013, 30, 565. [Google Scholar] [CrossRef] [PubMed]
- Haug, K.; Salek, R.M.; Conesa, P.; Hastings, J.; de Matos, P.; Rijnbeek, M.; Mahendraker, T.; Williams, M.; Neumann, S.; Rocca-Serra, P.; et al. MetaboLights—An open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 2013, 41, D781–D786. [Google Scholar] [CrossRef] [PubMed]
- Reem, N.T.; Chen, H.Y.; Hur, M.; Zhao, X.; Wurtele, E.S.; Li, X.; Li, L.; Zabotina, O. Comprehensive transcriptome analyses correlated with untargeted metabolome reveal differentially expressed pathways in response to cell wall alterations. Plant Mol. Biol. 2018, 96, 509–529. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing Real-Time PCR Data by the Comparative C(T) Method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Raw Base (bp) | Clean Base (bp) | Clean Read Number | Clean Read Rate (%) | Raw Q30 Base Rate | Clean Q30 Base Rate |
|---|---|---|---|---|---|---|
| A. venetum | 116,453,678,200 | 114,264,349,800 | 763,459,276 | 98.12 | 92.16 | 92.75 |
| A. hendersonii | 120,605,812,500 | 117,392,457,300 | 782,616,382 | 97.34 | 91.92 | 92.11 |
| Genome Characters | A. venetum | A. hendersonii | ||
|---|---|---|---|---|
| Contig | Scaffold | Contig | Scaffold | |
| Total length (bp) | 322,394,863 | 377,590,884 | 199,859,654 | 236,656,356 |
| Total number | 1250,389 | 876,453 | 741,647 | 303,264 |
| Max length (bp) | 6632 | 70,393 | 8521 | 110,436 |
| N50 length (bp) | 310 | 1225 | 457 | 4667 |
| N90 length (bp) | 132 | 150 | 114 | 217 |
| Genome Characters | A. venetum | A. hendersonii |
|---|---|---|
| K-mer number | 93,121,402,015 | 93,494,221,128 |
| K-mer depth | 400 | 400 |
| Genome size (Mbp) | 232.80 | 233.74 |
| Revised genome size (Mbp) | 224.36 | 228.21 |
| Heterozygosity ratio (%) | 0.67 | 0.46 |
| Metabolites | Retention Time (min) | m/z | Formula | A. venetum vs. A. hendersonii log2 (FC, Fold Change) |
|---|---|---|---|---|
| 1. Flavones | ||||
| Luteolin | 10.377 | 267.031 | C15H10O6 | - |
| Apigenin | 3.338 | 271.060 | C15H10O5 | - |
| 2. Flavonols | ||||
| Rutin | 3.430 | 591.138 | C27H30O16 | 1.788 |
| Hyperin | 3.313 | 465.103 | C21H20O12 | 3.452 |
| Isoquercitrin | 3.314 | 463.087 | C21H20O12 | 2.955 |
| Quercetin | 3.460 | 303.050 | C15H10O7 | 7.967 |
| Kaempferol | 4.066 | 285.039 | C15H10O6 | 4.030 |
| Tamarixetin | 3.550 | 363.07 | C16H14O7 | - |
| 3. Flavanones | ||||
| Hesperidin | 3.632 | 591.172 | C28H34O15 | −2.302 |
| 4. Isoflavanone | ||||
| Trifolin | 3.479 | 429.083 | C21H20O11 | - |
| 5. Flavanols | ||||
| Epigallocatechin | 2.463 | 307.081 | C15H14O7 | −0.144 |
| Catechin | 2.977 | 883.210 | C45H38O19 | −1.001 |
| Epicatechin | 3.763 | 271.062 | C15H14O6 | - |
| 6. Anthocyanidins | ||||
| Cyanidin | 3.216 | 287.055 | C43H42O22 | 1.952 |
| Procyanidin C1 | 3.097 | 867.215 | C15H10O6 | 0.959 |
| Procyanidin | 3.017 | 579.150 | C45H38O18 | - |
| Delphinidin | 3.313 | 303.050 | C15H10O7 | 0.118 |
| Pelargonidin | 11.853 | 611.049 | C15H11ClO5 | - |
| Malvidin | 11.268 | 347.033 | C17H15ClO7 | - |
| Peonidin | 10.826 | 381.038 | C16H13ClO6 | - |
| 7. Others | ||||
| Eriodictyol | 3.001 | 577.135 | C15H12O6 | −1.042 |
| Acacetin-7-O-rutinoside | 4.261 | 677.247 | C25H24O14 | 2.051 |
| Chrysoeriol 7-O-glucoside | 3.583 | 547.109 | C25H24O14 | 4.863 |
| Carthamin | 2.463 | 909.210 | C43H42O22 | - |
| Neocarthamin | 3.518 | 449.108 | C21H22O11 | −0.740 |
| Species | Hypersoside (mg/g Dry Weight) | Isoquercitrin (mg/g Dry Weight) | Quercetin (mg/g Dry Weight) | Rutin (mg/g Dry Weight) | Total Anthocyanins (mg/g Dry Weight) |
|---|---|---|---|---|---|
| A. venetum | 3.896 ± 0.043 | 5.254 ± 0.057 | 0.543 ± 0.036 | 3.653 ± 0.012 | 0.9017 ± 0.002 |
| A. hendersonii | 1.262 ± 0.025 | 1.718 ± 0.029 | 0.384 ± 0.015 | 2.275 ± 0.015 | 0.4133 ± 0.002 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, G.; Chen, P.; Chen, J.; Chen, K.; Wang, X.; Abubakar, A.S.; Liu, N.; Yu, C.; Zhu, A. Genomic Survey, Transcriptome, and Metabolome Analysis of Apocynum venetum and Apocynum hendersonii to Reveal Major Flavonoid Biosynthesis Pathways. Metabolites 2019, 9, 296. https://doi.org/10.3390/metabo9120296
Gao G, Chen P, Chen J, Chen K, Wang X, Abubakar AS, Liu N, Yu C, Zhu A. Genomic Survey, Transcriptome, and Metabolome Analysis of Apocynum venetum and Apocynum hendersonii to Reveal Major Flavonoid Biosynthesis Pathways. Metabolites. 2019; 9(12):296. https://doi.org/10.3390/metabo9120296
Chicago/Turabian StyleGao, Gang, Ping Chen, Jikang Chen, Kunmei Chen, Xiaofei Wang, Aminu Shehu Abubakar, Ning Liu, Chunming Yu, and Aiguo Zhu. 2019. "Genomic Survey, Transcriptome, and Metabolome Analysis of Apocynum venetum and Apocynum hendersonii to Reveal Major Flavonoid Biosynthesis Pathways" Metabolites 9, no. 12: 296. https://doi.org/10.3390/metabo9120296
APA StyleGao, G., Chen, P., Chen, J., Chen, K., Wang, X., Abubakar, A. S., Liu, N., Yu, C., & Zhu, A. (2019). Genomic Survey, Transcriptome, and Metabolome Analysis of Apocynum venetum and Apocynum hendersonii to Reveal Major Flavonoid Biosynthesis Pathways. Metabolites, 9(12), 296. https://doi.org/10.3390/metabo9120296