Metabolomics Test Materials for Quality Control: A Study of a Urine Materials Suite

 , ,
, ,  , , ,
, , ,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Interlaboratory Study Design
2.2. Sample Descriptions
- SRM 3667 [5] is a single pool of human urine collected from healthy male and female donors;
- SRM 3672 [6] is single pool of human urine collected from donors who smoked more than one pack of cigarettes a day;
- SRM 3673 [7] is single pool of human urine collected from non-smoker donors who were not exposed to secondhand cigarette smoke;
- SRM 3671 [8] is composed of three different pools, each collected from donors from a different exposure population: smokers who smoked at least one pack of cigarettes a day, nonsmokers exposed to secondhand cigarette smoke, and nonsmokers not exposed to cigarette smoke;
- The RGTM is a version of the SRM 3673 material with an additive. The additive does not produce a response in most analytical platforms and so this material serves as a functional replicate of SRM 3673.
2.3. Sample Preparation Protocols
2.3.1. NMR Protocol Summarized
2.3.2. LC-MS Protocol Summarized
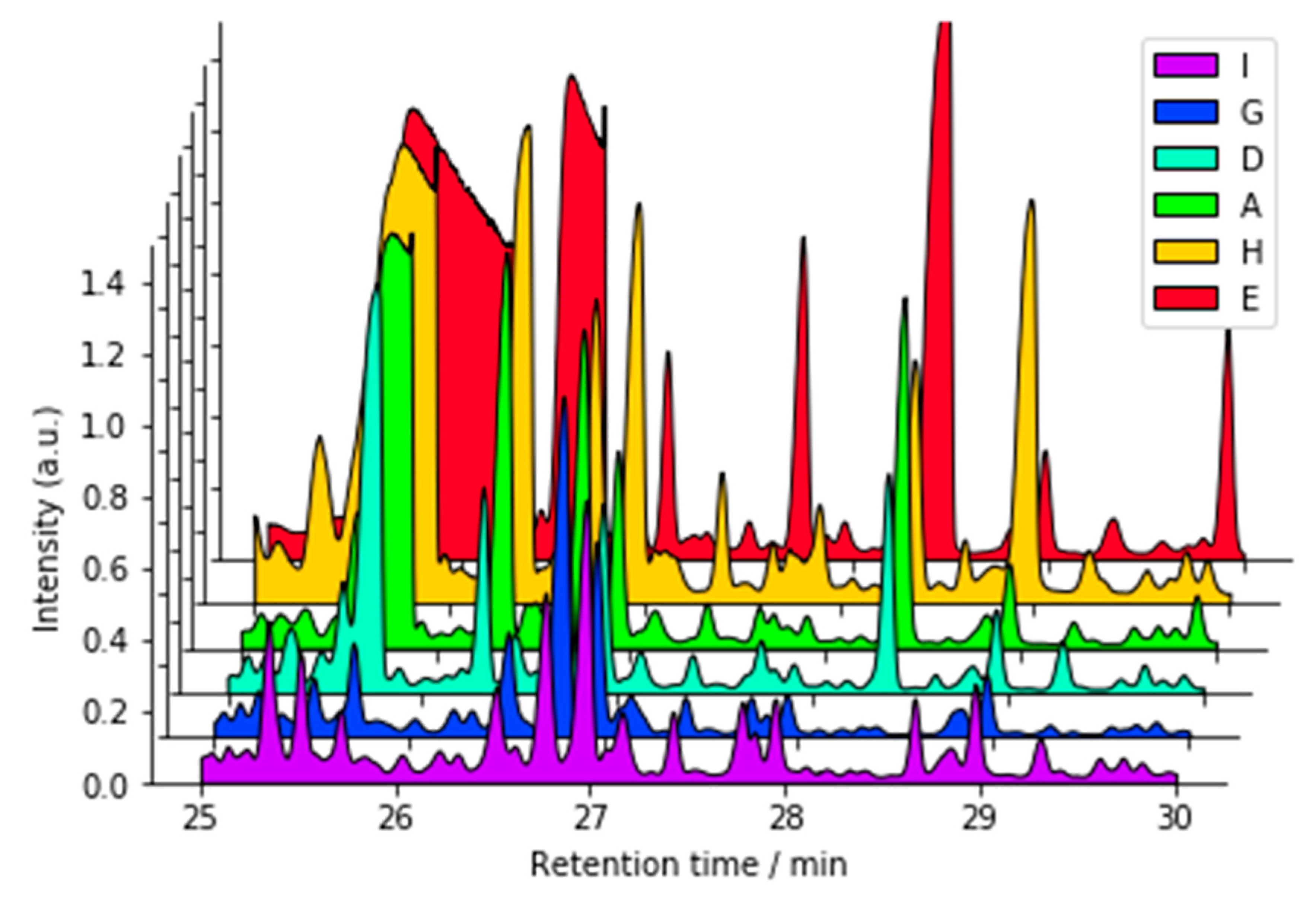
2.3.3. GC-MS Protocol Summarized
2.4. Chemometric Analysis
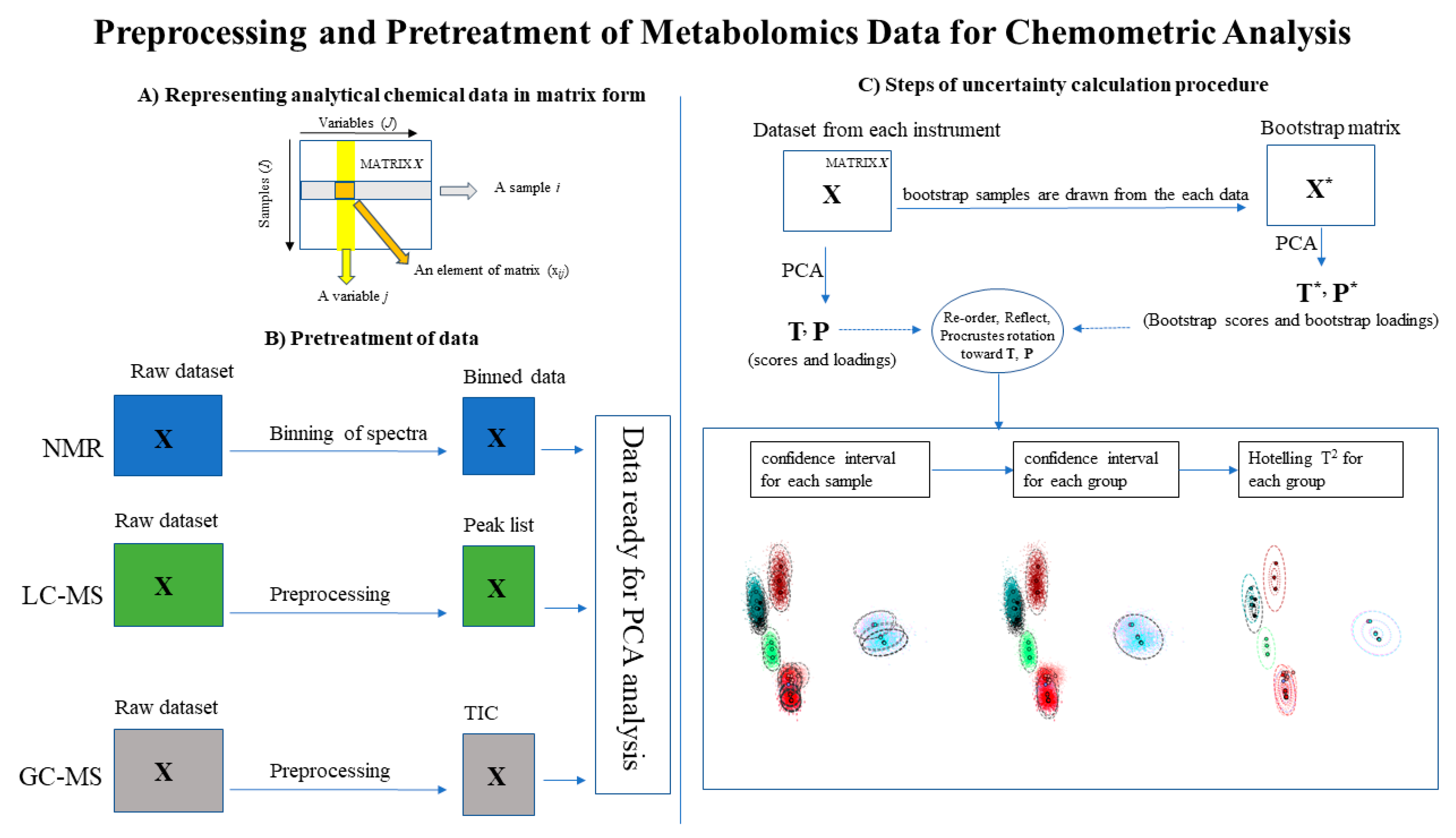
2.4.1. Preprocessing and Pretreatment
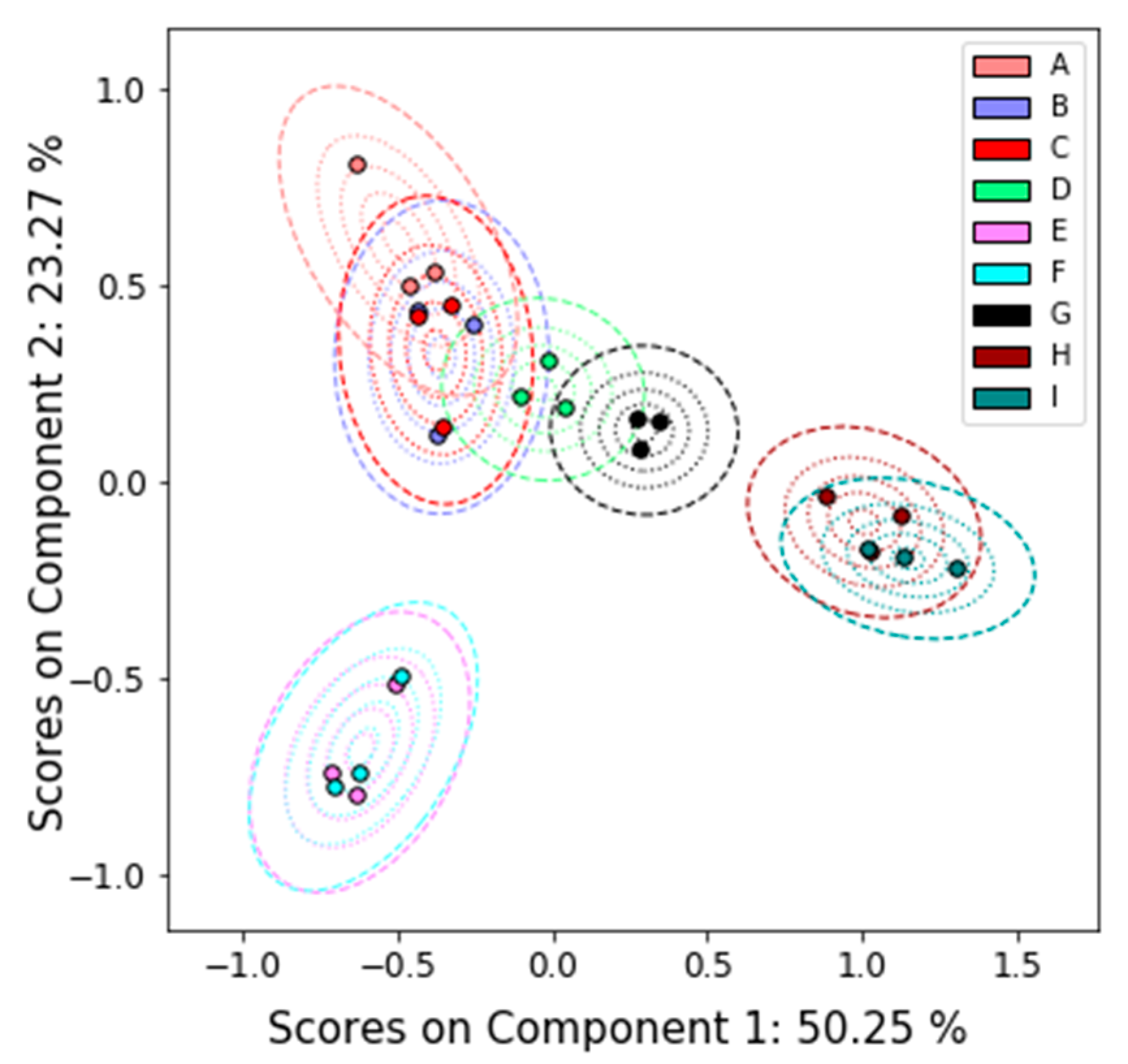
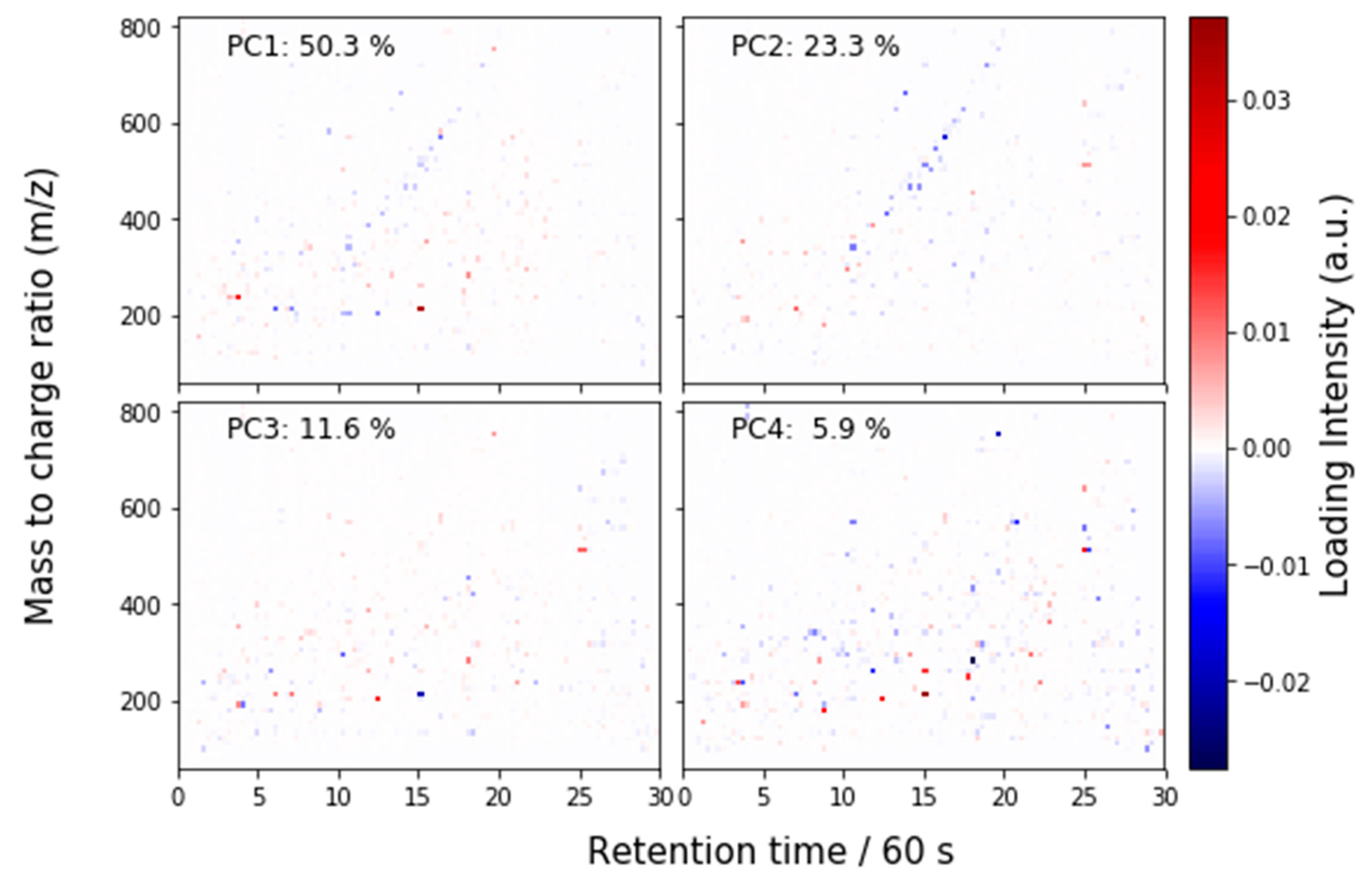
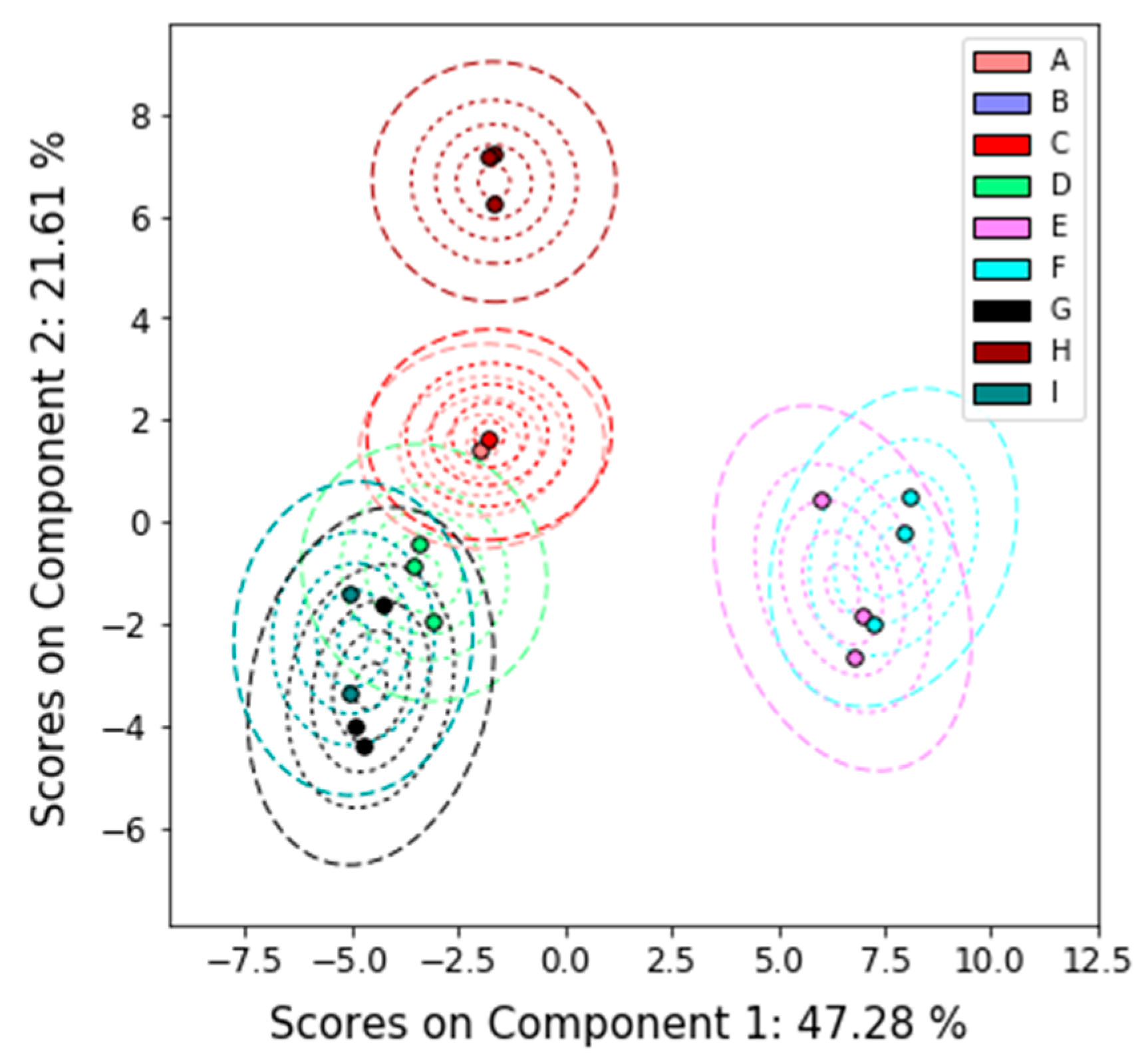
2.4.2. Principal Components Analysis
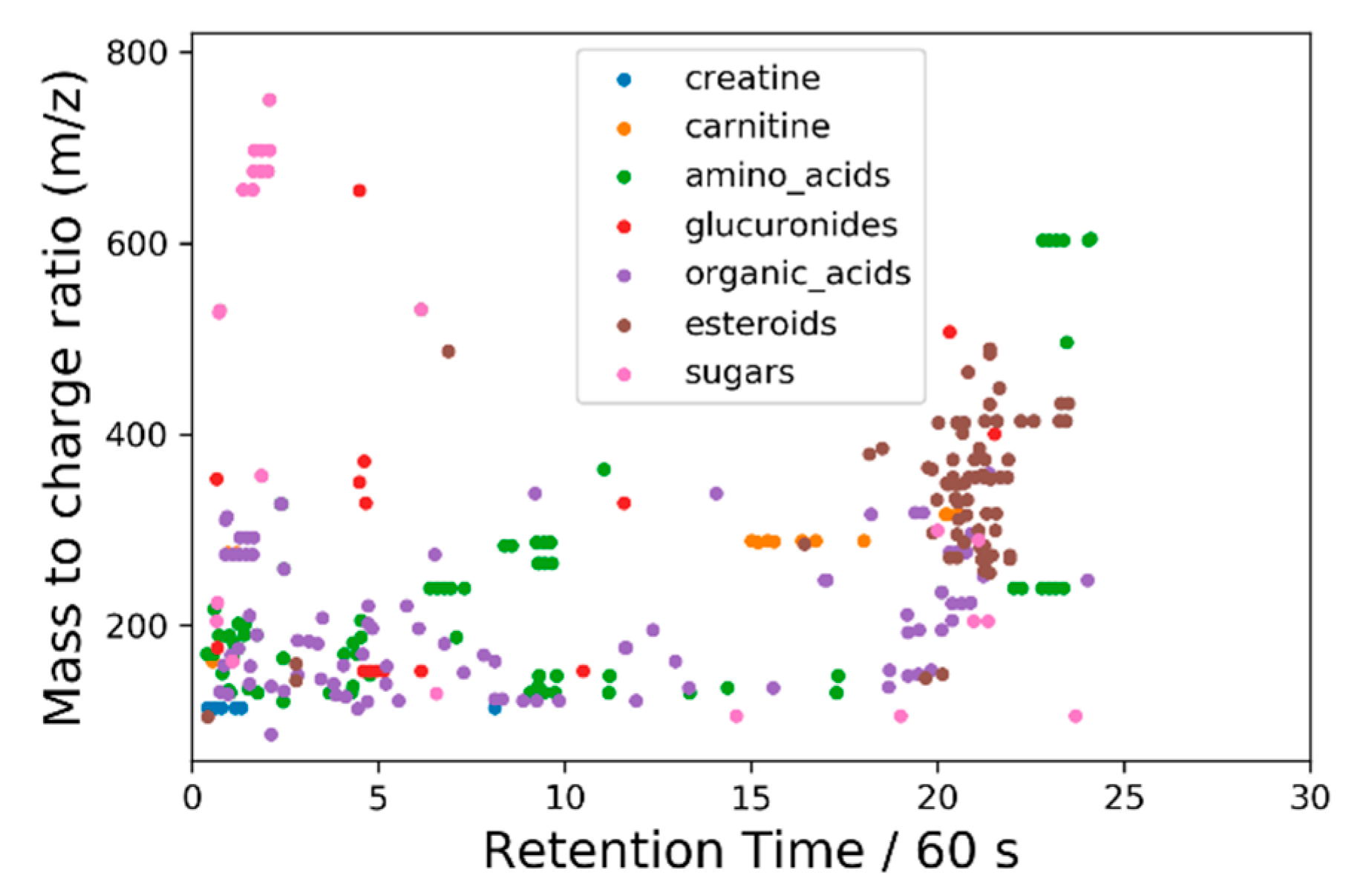
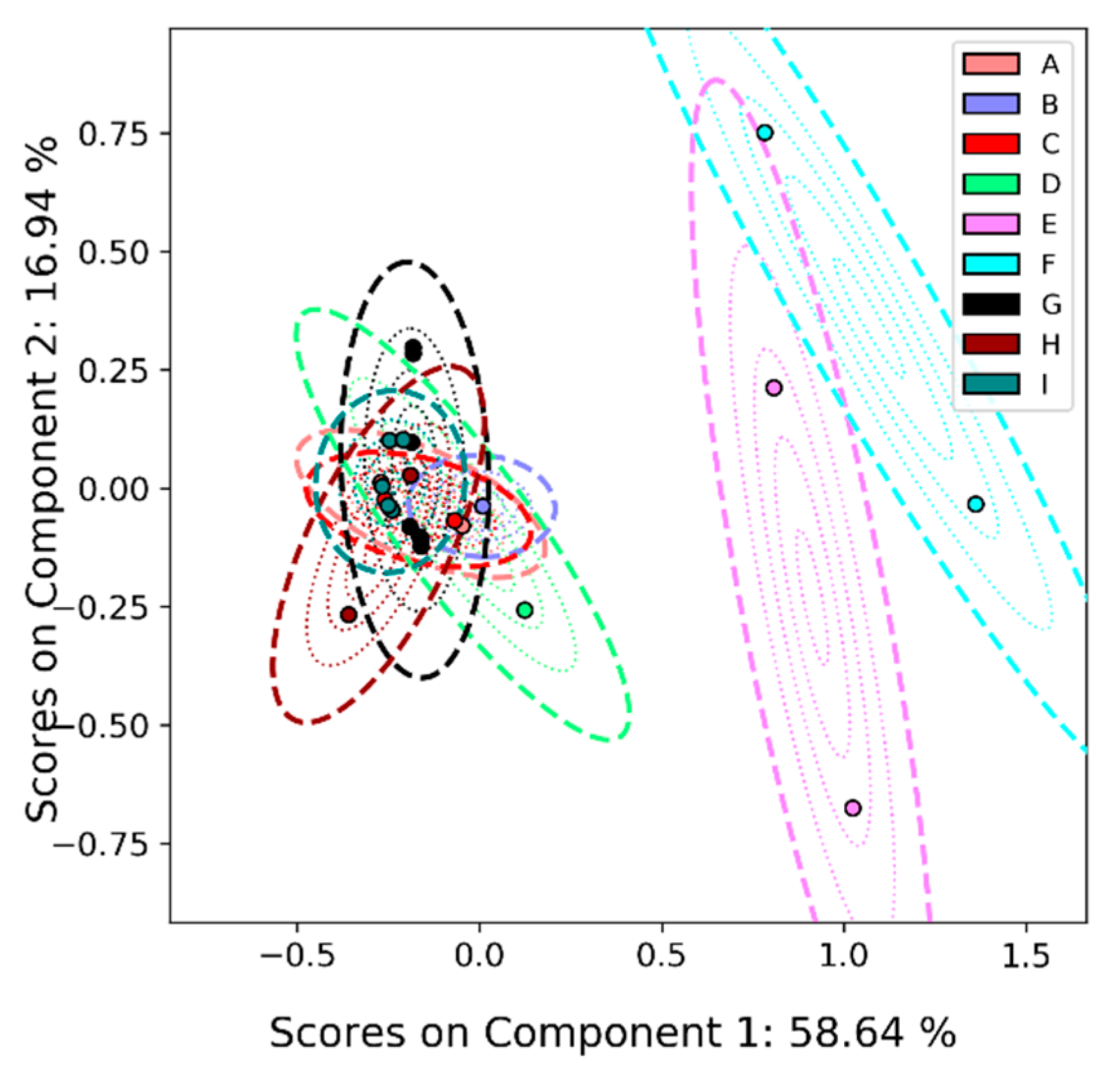
2.4.3. Principal Components Analysis on Feature List Data
3. Results
3.1. Overview
3.2. Detailed Results
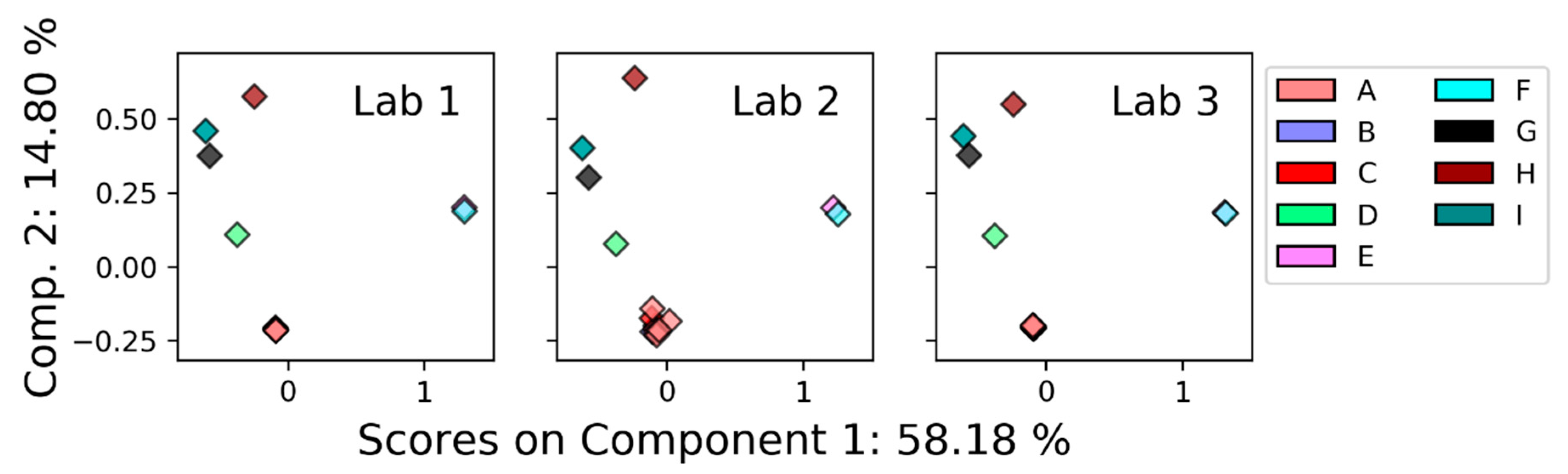
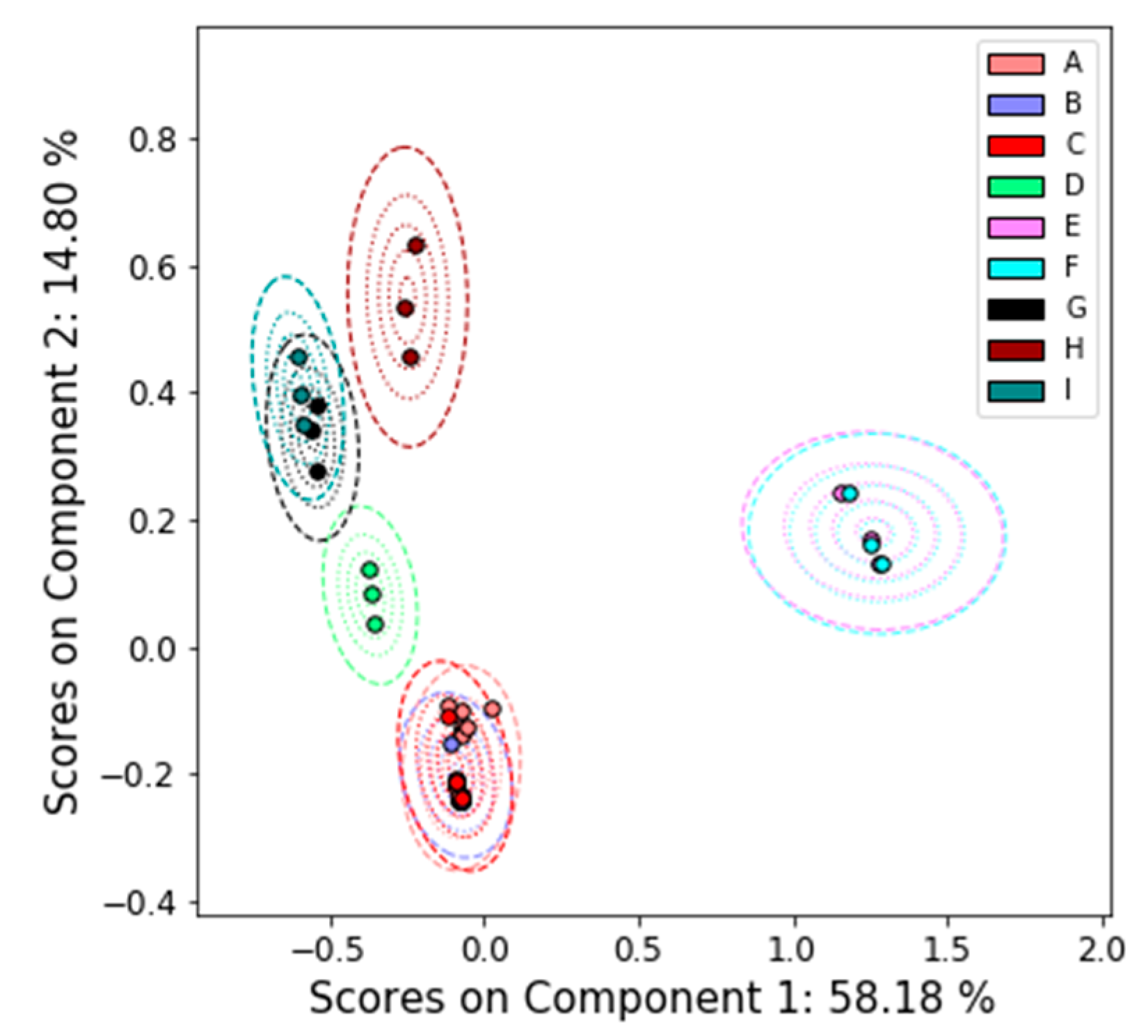
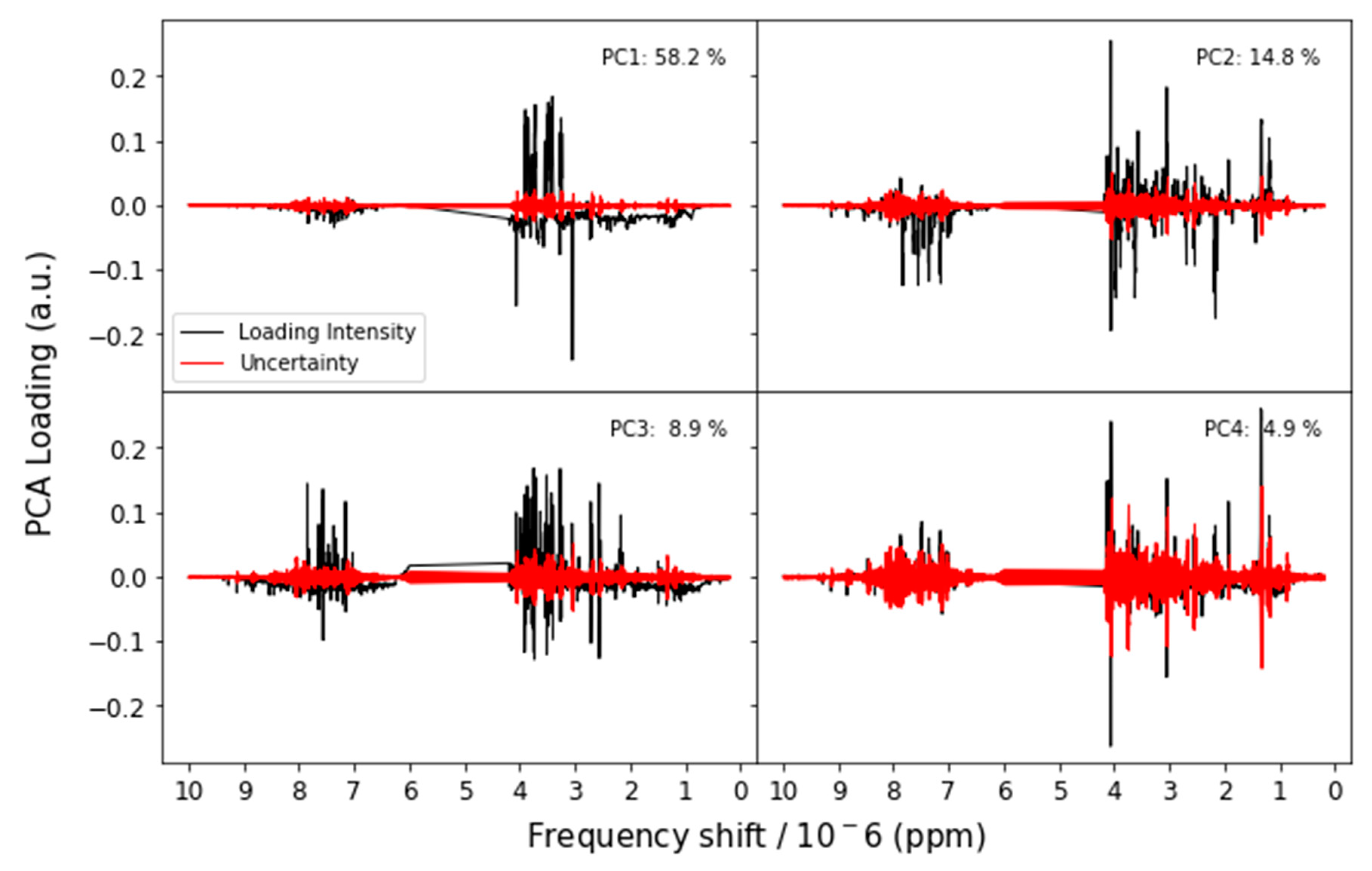
3.2.1. Nuclear Magnetic Resonance (NMR)
3.2.2. Liquid Chromatography–Mass Spectrometry (LC-MS)
3.2.3. Gas chromatography/Mass Spectrometry (GC-MS)
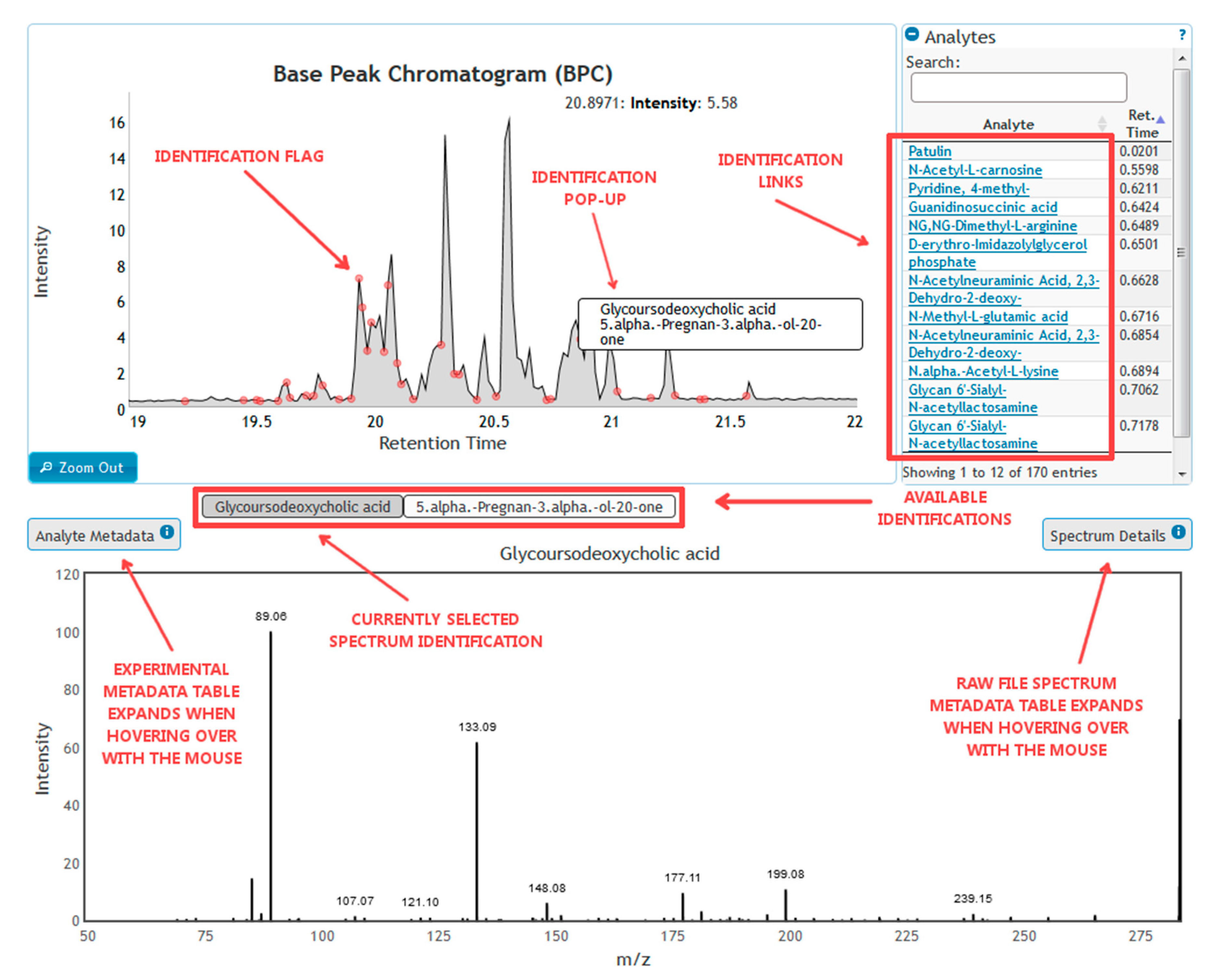
3.2.4. Data Dissemination
4. Discussion
4.1. Test Materials as A Reference Suite
4.2. Comparison between Platforms
4.3. Prospects and Availability of Test Materials
4.4. Contact
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments and Disclaimer
Author Copyright Statement
Conflicts of Interest
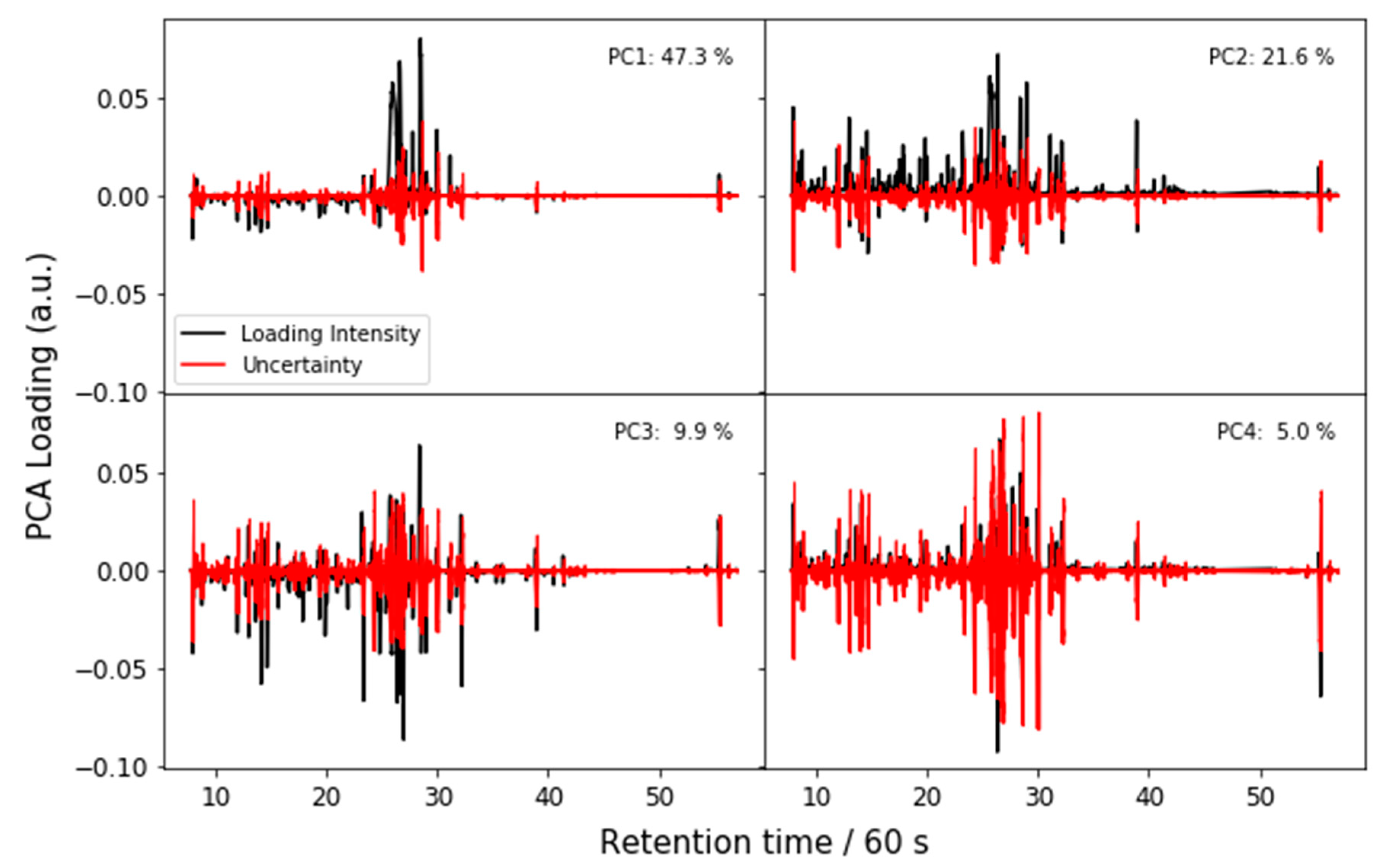
Appendix A. Uncertainty Analysis
Appendix A.1. Principal Components Analysis with Bootstrap Uncertainty (PCA-Boot)
Appendix A.2. PCA-Boot on LC-MS Peak List Data
Appendix A.3. PCA-Boot Detailed Procedure
- Sample randomly with replacement from the binned, normalized spectra in X (generate X*)
- Fit a Pareto scaler to the bootstrap sample X* (NMR only)
- Conduct PCA on the scaled X* (generate T* and P*)
- Scale X using the Pareto scaler (NMR only)
- Transform the scaled X into the X* PCA space (generate Tboot)
- Align Tboot with T
Appendix A.4. PCA-Boot on Peak Lists Detailed Procedure
- Conduct PCA on each facility’s peak list (calculate Ti and Pi)
- Choose a lab to use as the base lab (choose j)
- Align all Ti arrays with Tj (generate Ti,aligned)
- Map aligned scores into virtual peak lists (generate Xi,virtual)
- Conduct PCA and bootstrapping on concatenated virtual peak lists.
References
- Dunn, W.B.; Broadhurst, D.I.; Edison, A.; Guillou, C.; Viant, M.R.; Bearden, D.W.; Beger, R.D. Quality assurance and quality control processes: Summary of a metabolomics community questionnaire. Metabolomics 2017, 13, 539. [Google Scholar] [CrossRef]
- ISO Guide 33:2015: Reference Materials—Good Practice in Using Reference Materials; International Standards Organization: Geneva, Switzerland, 2015.
- May, W.E.; Parris, R.; Beck, C.; Fassett, J.; Greenberg, R.; Guenther, F.; Kramer, G.; Wise, S.; Gills, T.; Colbert, J. Special Publication 260-136: Definitions of Terms and Modes Used at NIST for Value-Assignment of Reference Materials for Chemical Measurements; National Institute of Standards and Technology: Gaithersburg, MD, USA; U.S. Department of Commerce: Washington, DC, USA, 2000; Volume 260, p. 136.
- Sheen, D.A.; Rocha, W.F.; Lippa, K.A.; Bearden, D.W. A scoring metric for multivariate data for reproducibility analysis using chemometric methods. Chemom. Intell. Lab. Syst. 2017, 162, 10–20. [Google Scholar] [CrossRef] [PubMed]
- SRM 3667; Creatinine in Frozen Human Urine; National Institute of Standards and Technology: Gaithersburg, MD, USA; U.S. Department of Commerce: Washington, DC, USA, 2017.
- SRM 3671; Nicotine Metabolites in Human Urine (Frozen); National Institute of Standards and Technology: Gaithersburg, MD, USA; U.S. Department of Commerce: Washington, DC, USA, 2015.
- SRM 3672; Organic Contaminants in Smokers’ Urine (Frozen); National Institute of Standards and Technology: Gaithersburg, MD, USA; U.S. Department of Commerce: Washington, DC, USA, 2015.
- SRM 3673; Organic Contaminants in Non-Smokers’ Urine (Frozen); National Institute of Standards and Technology: Gaithersburg, MD, USA; U.S. Department of Commerce: Washington, DC, USA, 2015.
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Simón-Manso, Y.; Lowenthal, M.S.; Kilpatrick, L.E.; Sampson, M.L.; Telu, K.H.; Rudnick, P.A.; Mallard, W.G.; Bearden, D.W.; Schock, T.B.; Tchekhovskoi, D.V.; et al. Metabolite Profiling of a NIST Standard Reference Material for Human Plasma (SRM 1950): GC-MS, LC-MS, NMR, and Clinical Laboratory Analyses, Libraries, and Web-Based Resources. Anal. Chem. 2013, 85, 11725–11731. [Google Scholar] [CrossRef] [PubMed]
- Telu, K.H.; Yan, X.; Wallace, W.E.; Stein, S.E.; Simon-Manso, Y. Analysis of human plasma metabolites across different liquid chromatography/mass spectrometry platforms: Cross-platform transferable chemical signatures. Rapid Commun. Mass Spectrom. 2016, 30, 581–593. [Google Scholar] [CrossRef]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J.K. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 2012, 8, 17–32. [Google Scholar] [CrossRef]
- Grob, K. Evaluation of capillary columns by separation number or plate number. J. Chromatogr. A 1981, 207, 291–297. [Google Scholar] [CrossRef]
- Chan, E.C.Y.; Pasikanti, K.K.; Nicholson, J.K. Global urinary metabolic profiling procedures using gas chromatography–mass spectrometry. Nat. Protoc. 2011, 6, 1483–1499. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Bro, R.; Smilde, A.K. Principal component analysis. Anal. Methods 2014, 6, 2812–2831. [Google Scholar] [CrossRef]
- Babamoradi, H.; Berg, F.V.D.; Rinnan, Å. Bootstrap based confidence limits in principal component analysis—A case study. Chemom. Intell. Lab. Syst. 2013, 120, 97–105. [Google Scholar] [CrossRef]
- Efron, B. Bootstrap Methods: Another Look at the Jackknife. Ann. Stat. 1979, 7, 1–26. [Google Scholar] [CrossRef]
- Milan, L.; Whittaker, J. Application of the Parametric Bootstrap to Models that Incorporate a Singular Value Decomposition. J. R. Stat. Soc. Ser. C Appl. Stat. 1995, 44, 31. [Google Scholar] [CrossRef]
- Simón-Manso, Y.; Yan, X.; Telu, K.H.; Mirokhin, Y.; Liang, Y.; Stein, S.E. Standardization and Harmonization of LC-MS Bioanalysis using Certified Reference Materials and Libraries of Recurrent Mass Spectra. In Proceedings of the 66th ASMS Conference on Mass Spectrometry and Allied Topics, San Diego, CA, USA, 3–7 June 2018. [Google Scholar]
- Simón-Manso, Y.; Marupaka, R.; Yan, X.; Liang, Y.; Telu, K.H.; Mirokhin, Y.; Stein, S.E. Mass Spectrometry Fingerprints of Small-Molecule Metabolites in Biofluids: Building a Spectral Library of Recurrent Spectra for Urine Analysis. Anal. Chem. 2019, 91, 12021–12029. [Google Scholar]
- Burke, M.C.; Mirokhin, Y.A.; Tchekhovskoi, D.V.; Markey, S.P.; Thompson, J.H.; Larkin, C.; Stein, S.E. The Hybrid Search: A Mass Spectral Library Search Method for Discovery of Modifications in Proteomics. J. Proteome Res. 2017, 16, 1924–1935. [Google Scholar] [CrossRef]
- Cooper, B.T.; Yan, X.; Simón-Manso, Y.; Tchekhovskoi, D.V.; Mirokhin, Y.A.; Stein, S.E. Hybrid Search: A Method for Identifying Metabolites Absent from Tandem Mass Spectrometry Libraries. Anal. Chem. 2019. [Google Scholar] [CrossRef]
- Moorthy, A.S.; Wallace, W.E.; Kearsley, A.J.; Tchekhovskoi, D.V.; Stein, S.E. Combining Fragment-Ion and Neutral-Loss Matching during Mass Spectral Library Searching: A New General Purpose Algorithm Applicable to Illicit Drug Identification. Anal. Chem. 2017, 89, 13261–13268. [Google Scholar] [CrossRef]
- Markley, J.L.; Brüschweiler, R.; Edison, A.S.; Eghbalnia, H.R.; Powers, R.; Raftery, D.; Wishart, D.S. The future of NMR-based metabolomics. Curr. Opin. Biotech. 2017, 43, 34–40. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A Web-Based Platform to Process Untargeted Metabolomic Data. Anal. Chem. 2012, 4, 5035–5039. [Google Scholar] [CrossRef]
- Gowda, H.; Ivanisevic, J.; Johnson, C.H.; Kurczy, M.E.; Benton, H.P.; Rinehart, D.; Nguyen, T.; Ray, J.; Kuehl, J.; Arevalo, B.; et al. Interactive XCMS Online: Simplifying Advanced Metabolomic Data Processing and Subsequent Statistical Analyses. Anal. Chem. 2014, 86, 6931–6939. [Google Scholar] [CrossRef]
- Pasikanti, K.K.; Ho, P.C.; Chan, E.C.Y. Gas chromatography/mass spectrometry in metabolic profiling of biological fluids. J. Chromatogr. B 2008, 871, 202–211. [Google Scholar] [CrossRef]
- Struck-Lewicka, W.; Kordalewska, M.; Bujak, R.; Mpanga, A.Y.; Markuszewski, M.; Jacyna, J.; Matuszewski, M.; Kaliszan, R.; Markuszewski, M.J. Urine metabolic fingerprinting using LC–MS and GC–MS reveals metabolite changes in prostate cancer: A pilot study. J. Pharmaceut. Biomed. 2015, 111, 351–361. [Google Scholar] [CrossRef]
- Blonder, N.; Orsburn, B.C.; Blonder, J.; Gonzalez, C.A. Visual Mass-Spec Share (vMS-Share): A New Public Web-Based Mass Spectrometry Visualization and Data Mining Repository. J. Proteom. Bioinf. 2019, 12, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.C.; Maillot, M.; Mazerolles, G.; Verdu, A.; Lyan, B.; Migne, C.; Defoort, C.; Canlet, C.; Junot, C.; Guillou, C.; et al. Can we trust untargeted metabolomics? Results of the metabo-ring initiative, a large-scale, multi-instrument inter-laboratory study. Metabolomics 2015, 11, 807–821. [Google Scholar] [PubMed]
- Cajka, T.; Smilowitz, J.T.; Fiehn, O. Validating Quantitative Untargeted Lipidomics Across Nine Liquid Chromatography–High-Resolution Mass Spectrometry Platforms. Anal. Chem. 2017, 89, 12360–12368. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lab | Distance from Lab | ||
|---|---|---|---|
| 1 | 2 | 3 | |
| 1 | 0.150 | 0.046 | |
| 2 | 0.150 | - | 0.169 |
| 3 | 0.046 | 0.169 | - |
| Lab | Number of Extracted Ions (Intensity > 5000 Counts) | Extracted Ions (RSD < 20% in All Samples) | Overlapping Extracted Ions with Lab | |||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||
| 1 | 28,818 | 13,867 | - | 12,511 | 8159 | 1225 |
| 2 | 42,781 | 12,563 | 12,511 | - | 7857 | 1141 |
| 3 | 56,656 | 10,332 | 8159 | 7857 | - | 3734 |
| 4 | 4947 | 4932 | 1225 | 1141 | 3734 | - |
| Lab | Total Number of Peaks Detected | Number of Identified Peaks | Identifications Overlapping |
|---|---|---|---|
| 1 | 71 to 130 | 50 to 93 | 12 to 26 |
| 2 | 227 to 268 | 62 to 95 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bearden, D.W.; Sheen, D.A.; Simón-Manso, Y.; Benner, B.A., Jr.; Rocha, W.F.C.; Blonder, N.; Lippa, K.A.; Beger, R.D.; Schnackenberg, L.K.; Sun, J.; et al. Metabolomics Test Materials for Quality Control: A Study of a Urine Materials Suite. Metabolites 2019, 9, 270. https://doi.org/10.3390/metabo9110270
Bearden DW, Sheen DA, Simón-Manso Y, Benner BA Jr., Rocha WFC, Blonder N, Lippa KA, Beger RD, Schnackenberg LK, Sun J, et al. Metabolomics Test Materials for Quality Control: A Study of a Urine Materials Suite. Metabolites. 2019; 9(11):270. https://doi.org/10.3390/metabo9110270
Chicago/Turabian StyleBearden, Daniel W., David A. Sheen, Yamil Simón-Manso, Bruce A. Benner, Jr., Werickson F. C. Rocha, Niksa Blonder, Katrice A. Lippa, Richard D. Beger, Laura K. Schnackenberg, Jinchun Sun, and et al. 2019. "Metabolomics Test Materials for Quality Control: A Study of a Urine Materials Suite" Metabolites 9, no. 11: 270. https://doi.org/10.3390/metabo9110270
APA StyleBearden, D. W., Sheen, D. A., Simón-Manso, Y., Benner, B. A., Jr., Rocha, W. F. C., Blonder, N., Lippa, K. A., Beger, R. D., Schnackenberg, L. K., Sun, J., Mehta, K. Y., Cheema, A. K., Gu, H., Marupaka, R., Nagana Gowda, G. A., & Raftery, D. (2019). Metabolomics Test Materials for Quality Control: A Study of a Urine Materials Suite. Metabolites, 9(11), 270. https://doi.org/10.3390/metabo9110270