Exposure of HepaRG Cells to Sodium Saccharin Underpins the Importance of Including Non-Hepatotoxic Compounds When Investigating Toxicological Modes of Action Using Metabolomics

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Methods
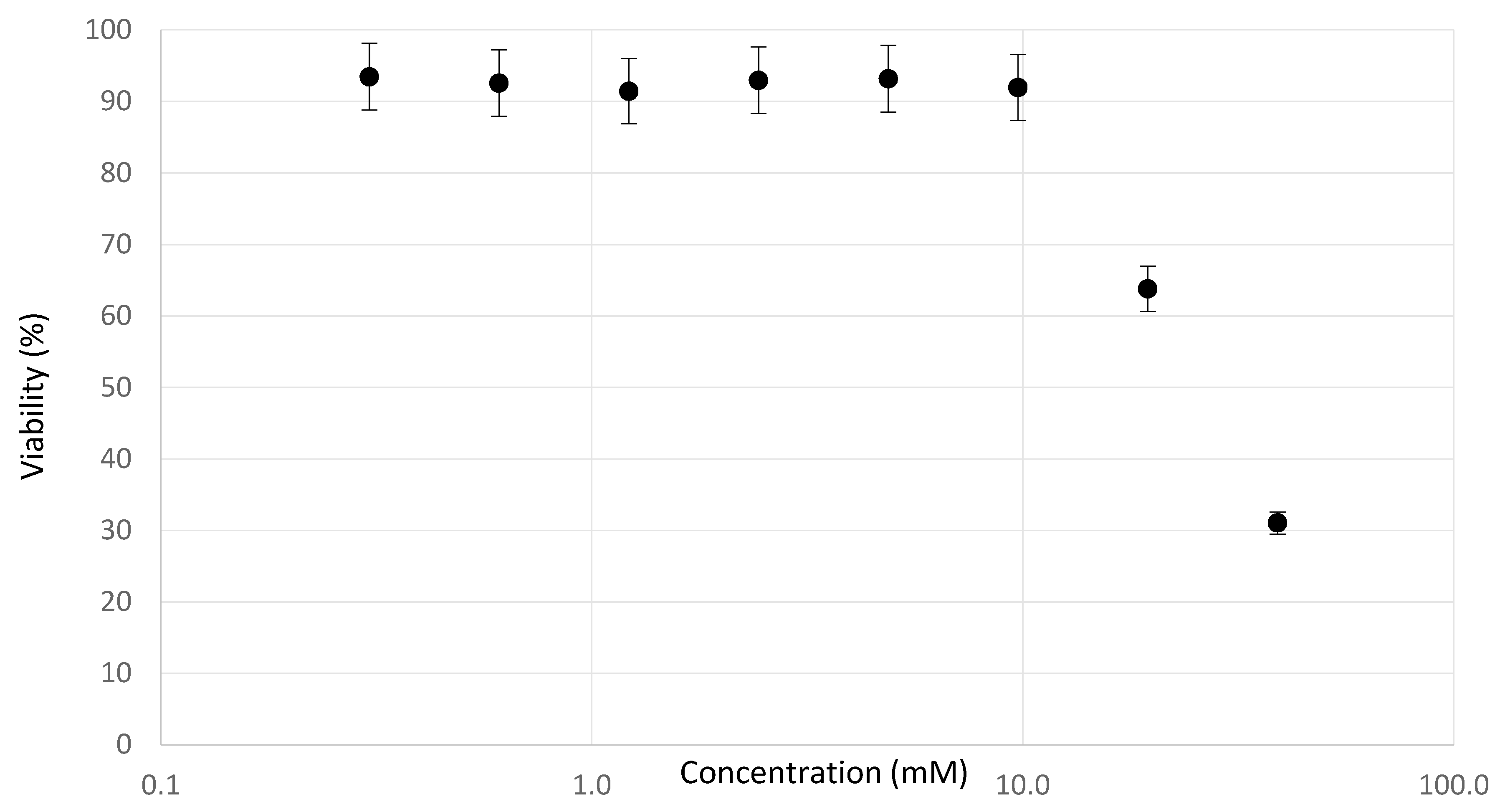
2.2. Determination of Testing Concentrations
2.3. Metabolomics Experiments
2.3.1. Seeding of the HepaRG® Cells and Exposure to Sodium Saccharin
2.3.2. Sample Preparation
2.3.3. LC-MS Analysis
2.3.4. Data Analysis
Data Quality Control
Data Pretreatment
Statistical Analysis
Metabolite Annotation
3. Results
3.1. Experimental Observations
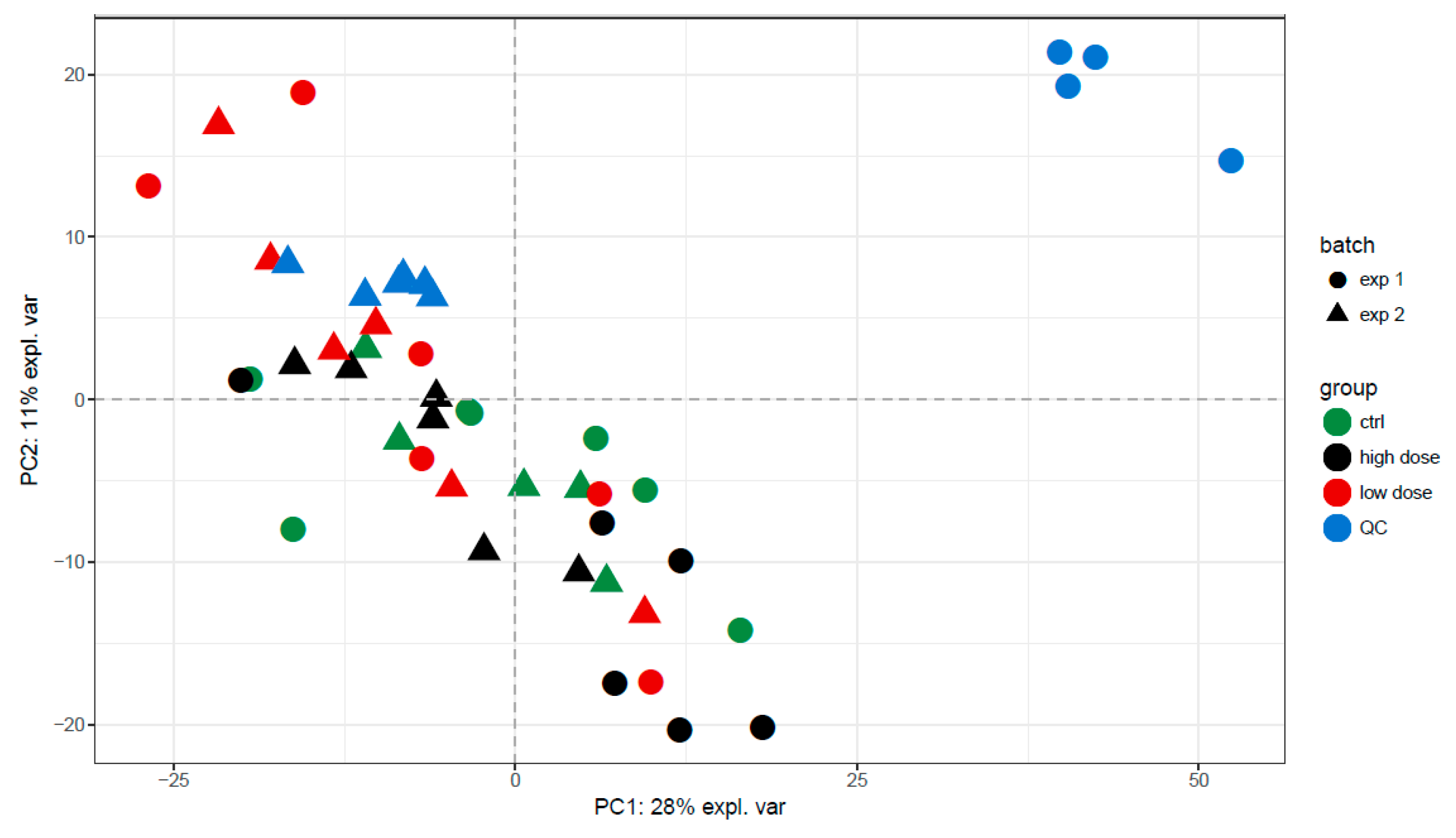
3.2. Data Quality
3.3. Selection of Potential Endogenous Markers of Exposure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. “Metabonomics”: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef]
- Balcke, G.U.; Kolle, S.N.; Kamp, H.; Bethan, B.; Looser, R.; Wagner, S.; Landsiedel, R.; van Ravenzwaay, B. Linking energy metabolism to dysfunctions in mitochondrial respiration—A metabolomics in vitro approach. Toxicol. Lett. 2011, 203, 200–209. [Google Scholar] [CrossRef]
- Ramirez, T.; Daneshian, M.; Kamp, H.; Bois, F.Y.; Clench, M.R.; Coen, M.; Donley, B.; Fischer, S.M.; Ekman, D.R.; Fabian, E.; et al. Metabolomics in toxicology and preclinical research. ALTEX 2013, 30, 209–225. [Google Scholar] [CrossRef]
- Broadhurst, D.I.; Kell, D.B. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2006, 2, 171–196. [Google Scholar] [CrossRef]
- Cuykx, M.; Rodrigues, R.M.; Laukens, K.; Vanhaecke, T.; Covaci, A. In vitro assessment of hepatotoxicity by metabolomics: A review. Arch. Toxicol. 2018, 92, 3007–3029. [Google Scholar] [CrossRef] [PubMed]
- Negro, F.; Mondardine, A.; Palmas, F. Hepatotoxicity of saccharin. N. Engl. J. Med. 1994, 330, 134–135. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine Toxnet: Hazardous Substance Database—Saccharin. Available online: http://toxnet.nlm.nih.gov (accessed on 1 October 2018).
- Cuykx, M.; Claes, L.; Rodrigues, R.M.; Vanhaecke, T.; Covaci, A. Metabolomics profiling of steatosis progression in HepaRG® cells using sodium valproate. Toxicol. Lett. 2018, 286, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-Z.; Lipsky, M.M.; Trump, B.F.; Hsu, I.-C. Neutral red (NR) assay for cell viability and xenobiotic-induced cytotoxicity in primary cultures of human and rat hepatocytes. Cell Biol. Toxicol. 1990, 6, 219–234. [Google Scholar] [CrossRef]
- Ates, G.; Vanhaecke, T.; Rogiers, V.; Rodrigues, R.M. Assaying cellular viability using the Neutral Red Uptake assay. Cell Viability Assays. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017; pp. 19–26. [Google Scholar]
- 1Hayton, S.; Maker, G.L.; Mullaney, I.; Trengove, R.D. Experimental design and reporting standards for metabolomics studies of mammalian cell lines. Cell. Mol. Life Sci. 2017, 74, 4421–4441. [Google Scholar] [CrossRef]
- Wu, H.; Southam, A.D.; Hines, A.; Viant, M.R. High-throughput tissue extraction protocol for NMR- and MS-based metabolomics. Anal. Biochem. 2008, 372, 204–212. [Google Scholar] [CrossRef]
- Cuykx, M.; Mortelé, O.; Rodrigues, R.M.; Vanhaecke, T.; Covaci, A. Optimisation of in vitro sample preparation for LC-MS metabolomics applications on HepaRG cell cultures. Anal. Methods 2017, 9, 3704–3712. [Google Scholar] [CrossRef]
- Dunn, W.B.; Wilson, I.D.; Nicholls, A.W.; Broadhurst, D. The importance of experimental design and QC samples in large-scale and MS-driven untargeted metabolomic studies of humans. Bioanalysis 2012, 4, 2249–2264. [Google Scholar] [CrossRef] [PubMed]
- Cuykx, M.; Negreira, N.; Beirnaert, C.; Van den Eede, N.; Rodrigues, R.; Vanhaecke, T.; Laukens, K.; Covaci, A. Tailored LC-MS analysis improves the coverage of the intracellular metabolome of HepaRG cells. J. Chromatogr. 2017, 1487, 168–178. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Smith, C.A.; Want, E.J.; Maille, G.O.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. ACS Publ. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Prince, J.T.; Marcotte, E.M. Chromatographic alignment of ESI-LC-MS proteomics data sets by ordered bijective interpolated warping. Anal. Chem. 2006, 78, 6140–6152. [Google Scholar] [CrossRef]
- Beirnaert, C.; Cuykx, M.; Bijtebier, S. MetaboMeeseeks: Helper functions for metabolomics analysis. R Package. 2019. version 0.1.10044. Available online: https://github.com/Beirnaert/MetaboMeeseeks (accessed on 25 September 2019).
- Wehrens, R.; Hageman, J.A.; van Eeuwijk, F.; Kooke, R.; Flood, P.J.; Wijnker, E.; Keurentjes, J.J.; Lommen, A.; van Eekelen, H.D.; Hall, R.D.; et al. Improved batch correction in untargeted MS-based metabolomics. Metabolomics 2016, 12, 88. [Google Scholar] [CrossRef]
- Pollard, K.S.; Dudoit, S.; Van der Laan, M.J. Multiple Testing Procedures: The multtest Package and Applications to Genomics. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: New York, NY, USA, 2005; pp. 249–271. [Google Scholar]
- Rohart, F.; Gautier, B.; Singh, A.; le Cao, K.-A. Le mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying small molecules via high resolution mass spectrometry: Communicating confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis: Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- Burtis, C.; Ashwood, E.; Bruns, D. Tietz Textbook of Clinical Chemistry and Molecular Diagnostics; Elsevier: St. Louis, MO, USA, 2006; ISBN 13 978-0-7216-0189-2. [Google Scholar]
- Sévin, D.C.; Stählin, J.N.; Pollak, G.R.; Kuehne, A.; Sauer, U. Global metabolic responses to salt stress in fifteen species. PLoS ONE 2016, 11, e0148888. [Google Scholar] [CrossRef]
- García-Cañaveras, J.C.; Castell, J.V.; Donato, M.T.; Lahoz, A. A metabolomics cell-based approach for anticipating and investigating drug-induced liver injury. Sci. Rep. 2016, 6, 27239. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, T.; Strigun, A.; Verlohner, A.; Huener, H.A.; Peter, E.; Herold, M.; Bordag, N.; Mellert, W.; Walk, T.; Spitzer, M.; et al. Prediction of liver toxicity and mode of action using metabolomics in vitro in HepG2 cells. Arch. Toxicol. 2017, 92, 839–906. [Google Scholar] [CrossRef] [PubMed]
- Cuykx, M.; Beirnaert, C.; Rodrigues, R.M.; Laukens, K.; Vanhaecke, T.; Covaci, A. Untargeted liquid chromatography-mass spectrometry metabolomics to assess drug-induced cholestatic features in HepaRG® cells. Toxicol. Appl. Pharmacol. 2019, 379, 114666. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Exposure | Non-Polar Positive | Non-Polar Negative | Polar Positive | Polar Negative |
|---|---|---|---|---|
| Low Dose | 0.51 | 0.55 | 0.59 | 0.17 |
| High Dose | 0.95 | 0.40 | 0.69 | 0.21 |
| Exposure | Non-Polar Positive | Non-Polar Negative | Polar Positive | Polar Negative | ||||
|---|---|---|---|---|---|---|---|---|
| R2 | Q2 | R2 | Q2 | R2 | Q2 | R2 | Q2 | |
| Low Dose | 0.01 | 0.01 | 0.1 | 0.06 | 0.22 | 0.14 | 0.21 | 0.05 |
| High Dose | 0.84 | 0.4 | 0.42 | 0.10 | 0.08 | 0.03 | 0.33 | 0.04 |
| Bosentan | Sodium Valproate | Sodium Saccharin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Time Frame | 24 h | 72 h | 24 h | 72 h | 72 h | ||||
| Concentration (µg/mL) | 23 | 230 | 9.5 | 95 | 230 | 2300 | 66.5 | 665 | 1000 |
| Acetylcholine | |||||||||
| Acetylspermidine | |||||||||
| Aminergic Oligopeptides | |||||||||
| Carnitine | |||||||||
| citric-acid N-sugar | |||||||||
| Choline | |||||||||
| Cholesterol Sulfate | |||||||||
| Creatine | |||||||||
| diacetylspermidine | |||||||||
| GTP | |||||||||
| Isoputreanine | |||||||||
| Methylbutyryl Carnitine | |||||||||
| Methylhydroxylysine | |||||||||
| Nucleotides | |||||||||
| Ornithine | |||||||||
| Pantothenic Acid | |||||||||
| Phosphocholine | |||||||||
| Phosphorylated Metabolites | |||||||||
| Phosphorylethanolamine | |||||||||
| Putrescine | |||||||||
| SAM | |||||||||
| Spermidine | |||||||||
| Taurine | |||||||||
| Trimethylammonium Butanoic Acid | |||||||||
| UDP Glucuronic Acid | |||||||||
| Bile Acids | |||||||||
| Ceramide | |||||||||
| Ceramide, Derivative | |||||||||
| Diacylglycerol | |||||||||
| Glycosfingolipid | |||||||||
| LPE 18:1 | |||||||||
| PC | |||||||||
| PE (non PUFA) | |||||||||
| PE (PUFA) | |||||||||
| PE (P) | |||||||||
| PS | |||||||||
| Sfingomyelin | |||||||||
| Triacylglycerol (O) | |||||||||
| Triacylglycerol (>50, PUFA) | |||||||||
| Triacylglycerol (>50, non PUFA) | |||||||||
| Triacylglycerol (<50) | |||||||||
| Colour | ||||||
|---|---|---|---|---|---|---|
| Number of Lipid Species | >3 | 1–3 | 0 | 1–3 | 4–10 | >10 |
| Signal Abundance | Lower | Lower | N/A | Higher | Higher | Higher |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuykx, M.; Beirnaert, C.; Rodrigues, R.M.; Laukens, K.; Vanhaecke, T.; Covaci, A. Exposure of HepaRG Cells to Sodium Saccharin Underpins the Importance of Including Non-Hepatotoxic Compounds When Investigating Toxicological Modes of Action Using Metabolomics. Metabolites 2019, 9, 265. https://doi.org/10.3390/metabo9110265
Cuykx M, Beirnaert C, Rodrigues RM, Laukens K, Vanhaecke T, Covaci A. Exposure of HepaRG Cells to Sodium Saccharin Underpins the Importance of Including Non-Hepatotoxic Compounds When Investigating Toxicological Modes of Action Using Metabolomics. Metabolites. 2019; 9(11):265. https://doi.org/10.3390/metabo9110265
Chicago/Turabian StyleCuykx, Matthias, Charlie Beirnaert, Robim Marcelino Rodrigues, Kris Laukens, Tamara Vanhaecke, and Adrian Covaci. 2019. "Exposure of HepaRG Cells to Sodium Saccharin Underpins the Importance of Including Non-Hepatotoxic Compounds When Investigating Toxicological Modes of Action Using Metabolomics" Metabolites 9, no. 11: 265. https://doi.org/10.3390/metabo9110265
APA StyleCuykx, M., Beirnaert, C., Rodrigues, R. M., Laukens, K., Vanhaecke, T., & Covaci, A. (2019). Exposure of HepaRG Cells to Sodium Saccharin Underpins the Importance of Including Non-Hepatotoxic Compounds When Investigating Toxicological Modes of Action Using Metabolomics. Metabolites, 9(11), 265. https://doi.org/10.3390/metabo9110265