Creating a Reliable Mass Spectral–Retention Time Library for All Ion Fragmentation-Based Metabolomics

 , , , ,
, , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
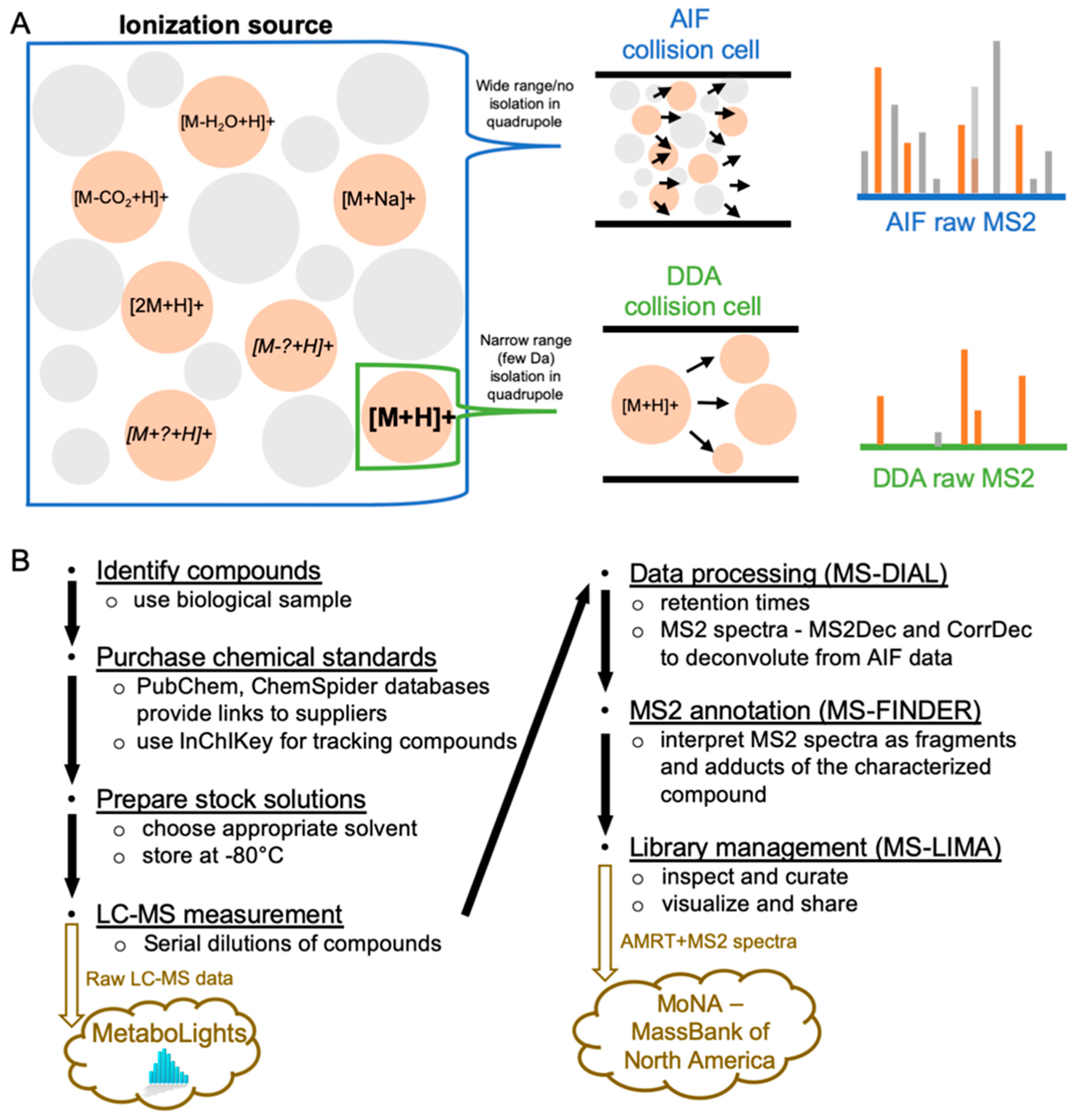
1. Introduction
2. Results and Discussion
2.1. Selection of Compounds for the Library
2.2. LC–MS Acquisition of the Chemical Standard Spectra
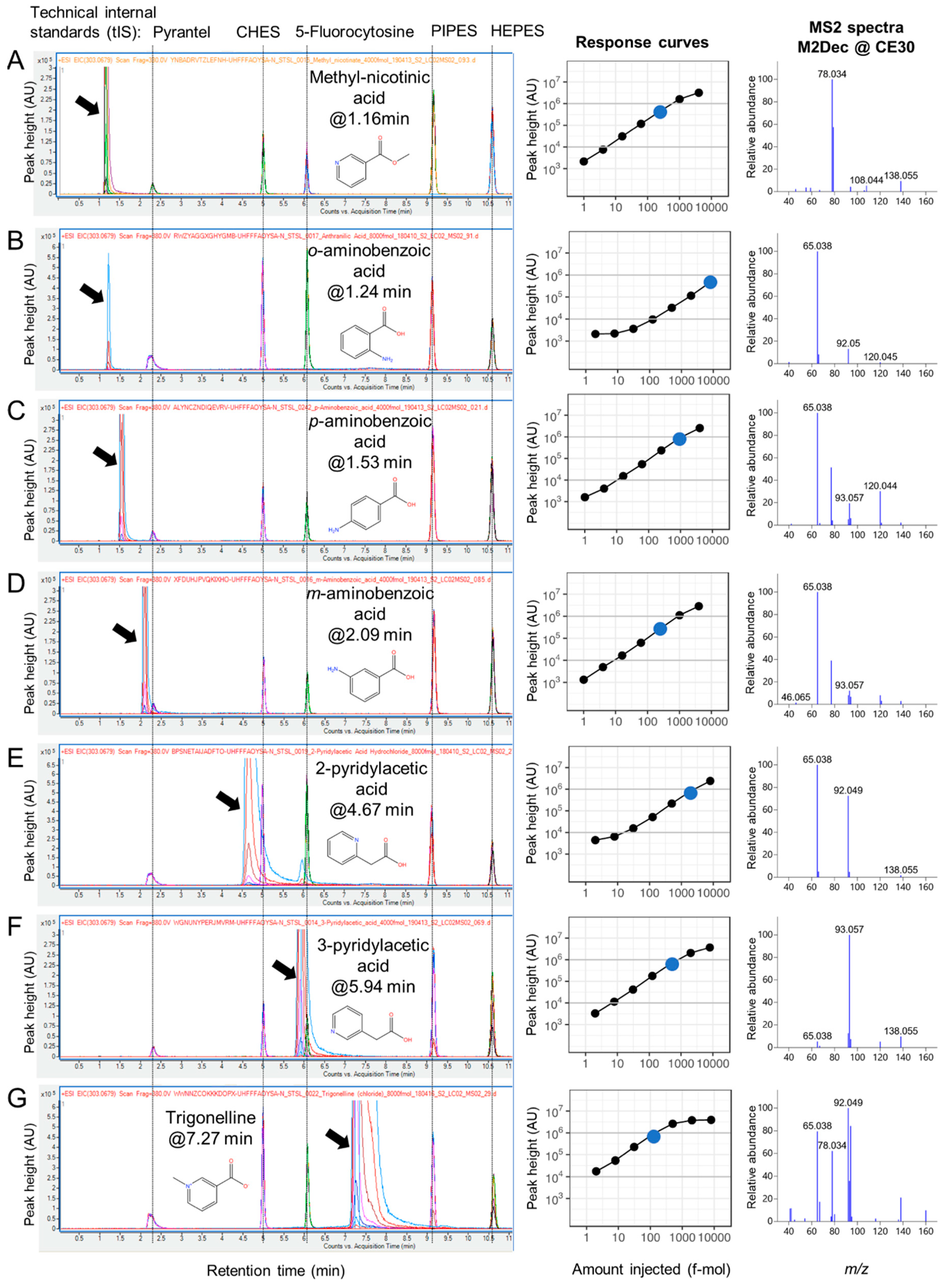
2.3. RT Characterization and Verification using Technical Internal Standards (tIS)
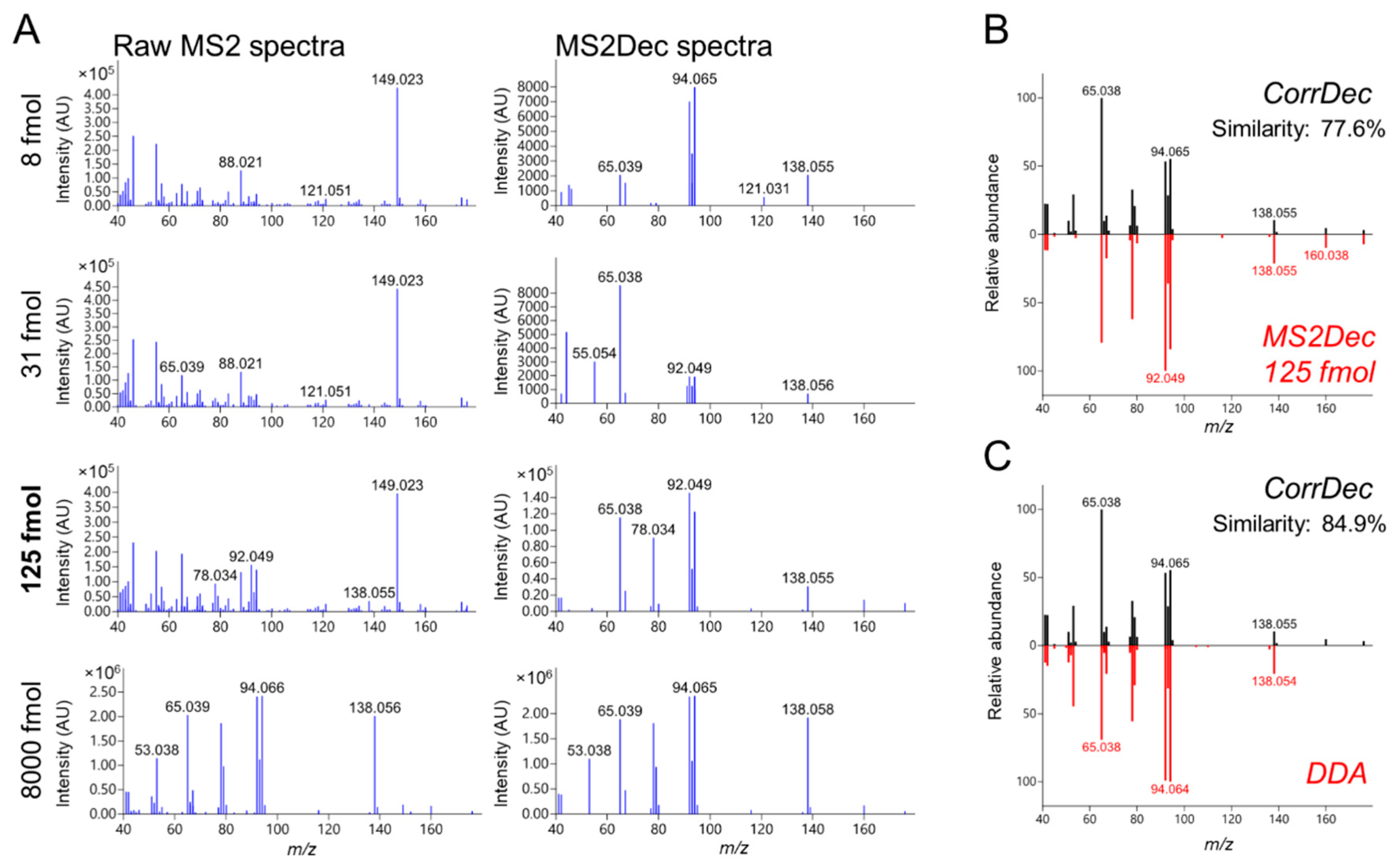
2.4. MS2 Spectra Deconvolution and Annotation of Major Ions Using AIF Data
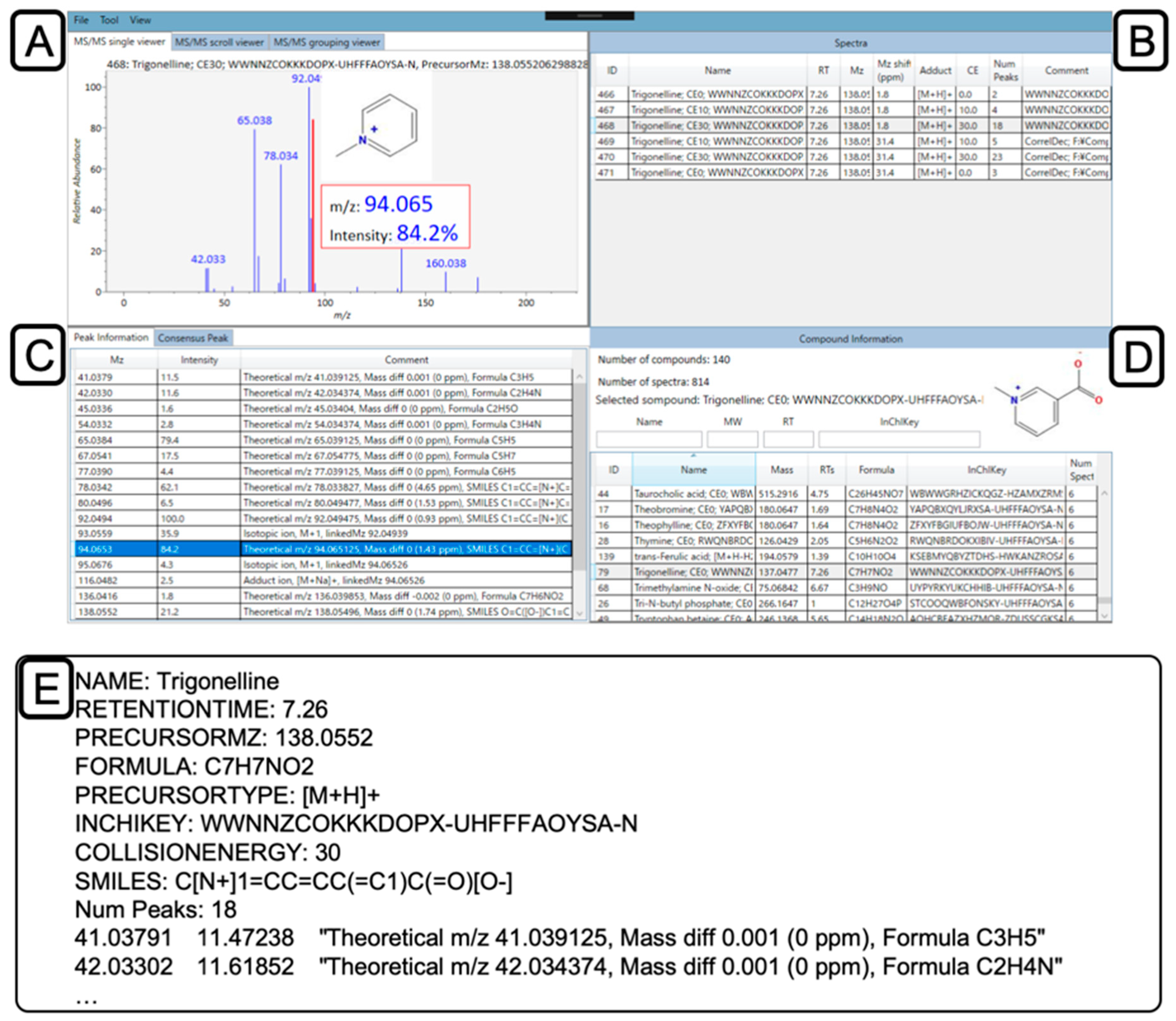
2.5. Confirmation and Curation of MS2 Spectra using MS-LIMA
2.6. Library Application for Human Urine Study and Limitations
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Compound Preparation for Analysis
4.3. Data Acquisition
4.4. Data Analysis
4.5. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rinschen, M.M.; Ivanisevic, J.; Giera, M.; Siuzdak, G. Identification of bioactive metabolites using activity metabolomics. Nat. Rev. Mol. Cell Biol. 2019, 20, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Fiehn, O. Toward merging untargeted and targeted methods in mass spectrometry-based metabolomics and lipidomics. Anal. Chem. 2016, 88, 524–545. [Google Scholar] [CrossRef] [PubMed]
- Chamkasem, N.; Ollis, L.W.; Harmon, T.; Lee, S.; Mercer, G. Analysis of 136 pesticides in avocado using a modified QuEChERS method with LC-MS/MS and GC-MS/MS. J. Agric. Food Chem. 2013, 61, 2315–2329. [Google Scholar] [CrossRef]
- Chaleckis, R.; Meister, I.; Zhang, P.; Wheelock, C.E. Challenges, progress and promises of metabolite annotation for LC-MS-based metabolomics. Curr. Opin. Biotechnol. 2019, 55, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Chen, Y.; Subramanian, R. Comparison of information-dependent acquisition, SWATH, and MS(All) techniques in metabolite identification study employing ultrahigh-performance liquid chromatography-quadrupole time-of-flight mass spectrometry. Anal. Chem. 2014, 86, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Nikolskiy, I.; Mahieu, N.G.; Chen, Y.J.; Tautenhahn, R.; Patti, G.J. An untargeted metabolomic workflow to improve structural characterization of metabolites. Anal. Chem. 2013, 85, 7713–7719. [Google Scholar] [CrossRef]
- Wang, R.; Yin, Y.; Zhu, Z.J. Advancing untargeted metabolomics using data-independent acquisition mass spectrometry technology. Anal. Bioanal. Chem. 2019, 411, 4349–4357. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Li, H.; Cai, Y.; Guo, Y.; Chen, F.; Zhu, Z.J. MetDIA: Targeted metabolite extraction of multiplexed MS/MS spectra generated by data-independent acquisition. Anal. Chem. 2016, 88, 8757–8764. [Google Scholar] [CrossRef]
- Tada, I.; Chaleckis, R.; Tsugawa, H.; Meister, I.; Zhang, P.; Lazarinis, N.; Dahlén, B.; Wheelock, C.E.; Arita, M. Correlation-based Deconvolution (CorrDec) to generate high quality MS2 spectra from data independent acquisition in multi-sample studies. Manuscript in preparation, in press.
- Domingo-Almenara, X.; Montenegro-Burke, J.R.; Guijas, C.; Majumder, E.L.; Benton, H.P.; Siuzdak, G. Autonomous METLIN-guided in-source fragment annotation for untargeted metabolomics. Anal. Chem. 2019, 91, 3246–3253. [Google Scholar] [CrossRef] [PubMed]
- Vinaixa, M.; Schymanski, E.L.; Neumann, S.; Navarro, M.; Salek, R.M.; Yanes, O. Mass spectral databases for LC/MS- and GC/MS-based metabolomics: State of the field and future prospects. TrAC Trends Anal. Chem. 2016, 78, 23–35. [Google Scholar] [CrossRef]
- Naz, S.; Gallart-Ayala, H.; Reinke, S.N.; Mathon, C.; Blankley, R.; Chaleckis, R.; Wheelock, C.E. Development of a liquid chromatography-high resolution mass spectrometry metabolomics method with high specificity for metabolite identification using all ion fragmentation acquisition. Anal. Chem. 2017, 89, 7933–7942. [Google Scholar] [CrossRef] [PubMed]
- Bruderer, T.; Varesio, E.; Hidasi, A.O.; Duchoslav, E.; Burton, L.; Bonner, R.; Hopfgartner, G. Metabolomic spectral libraries for data-independent SWATH liquid chromatography mass spectrometry acquisition. Anal. Bioanal. Chem. 2018, 410, 1873–1884. [Google Scholar] [CrossRef]
- Sentandreu, E.; Peris-Díaz, M.D.; Sweeney, S.R.; Chiou, J.; Muñoz, N.; Tiziani, S. A survey of orbitrap all ion fragmentation analysis assessed by an R MetaboList package to study small-molecule metabolites. Chromatographia 2018, 81, 981–994. [Google Scholar] [CrossRef]
- GitHub Repository for MS-LIMA. Available online: https://github.com/tipputa/MS-LIMA-Standard (accessed on 10 June 2019).
- Dunn, W.B.; Erban, A.; Weber, R.J.M.; Creek, D.J.; Brown, M.; Breitling, R.; Hankemeier, T.; Goodacre, R.; Neumann, S.; Kopka, J.; et al. Mass appeal: Metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics 2012, 9, 44–66. [Google Scholar] [CrossRef]
- Domingo-Almenara, X.; Montenegro-Burke, J.R.; Benton, H.P.; Siuzdak, G. Annotation: A computational solution for streamlining metabolomics analysis. Anal. Chem. 2018, 90, 480–489. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 2016, 44, D1214–D1219. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Pence, H.E.; Williams, A. ChemSpider: An online chemical information resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Dittmar, P.G.; Stobaugh, R.E.; Watson, C.E. The chemical abstracts service chemical registry system. I. General design. J. Chem. Inf. Comput. Sci. 1976, 16, 111–121. [Google Scholar] [CrossRef]
- Heller, S.R.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC international chemical identifier. J. Cheminf. 2015, 7, 23. [Google Scholar] [CrossRef]
- Szöcs, E.; Münch, D.; Ranke, J. {webchem}: Retrieve chemical information from the web. Zenodo 2015. [Google Scholar] [CrossRef]
- Wohlgemuth, G.; Haldiya, P.K.; Willighagen, E.; Kind, T.; Fiehn, O. The chemical translation service—A web-based tool to improve standardization of metabolomic reports. Bioinformatics 2010, 26, 2647–2648. [Google Scholar] [CrossRef]
- Kärkkäinen, O.; Lankinen, M.A.; Vitale, M.; Jokkala, J.; Leppänen, J.; Koistinen, V.; Lehtonen, M.; Giacco, R.; Rosa-Sibakov, N.; Micard, V.; et al. Diets rich in whole grains increase betainized compounds associated with glucose metabolism. Am. J. Clin. Nutr. 2018, 108, 971–979. [Google Scholar] [CrossRef]
- Chaleckis, R.; Naz, S.; Meister, I.; Wheelock, C.E. LC-MS-Based metabolomics of biofluids using all-ion fragmentation (AIF) acquisition. In Clinical Metabolomics: Methods and Protocols; Giera, M., Ed.; Humana Press: New York, NY, USA, 2018; Volume 1730, pp. 45–58. [Google Scholar]
- Pozo, O.J.; Van Eenoo, P.; Deventer, K.; Delbeke, F.T. Ionization of anabolic steroids by adduct formation in liquid chromatography electrospray mass spectrometry. J. Mass Spectrom. 2007, 42, 497–516. [Google Scholar] [CrossRef]
- Pluskal, T.; Nakamura, T.; Villar-Briones, A.; Yanagida, M. Metabolic profiling of the fission yeast S. pombe: Quantification of compounds under different temperatures and genetic perturbation. Mol. BioSyst. 2010, 6, 182–198. [Google Scholar] [CrossRef]
- Liigand, P.; Kaupmees, K.; Haav, K.; Liigand, J.; Leito, I.; Girod, M.; Antoine, R.; Kruve, A. Think negative: Finding the best electrospray ionization/MS mode for your analyte. Anal. Chem. 2017, 89, 5665–5668. [Google Scholar] [CrossRef]
- Abate-Pella, D.; Freund, D.M.; Ma, Y.; Simón-Manso, Y.; Hollender, J.; Broeckling, C.D.; Huhman, D.V.; Krokhin, O.V.; Stoll, D.R.; Hegeman, A.D.; et al. Retention projection enables accurate calculation of liquid chromatographic retention times across labs and methods. J. Chromatogr. A 2015, 1412, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Creek, D.J.; Barrett, M.P.; Blackburn, G.; Watson, D.G. Evaluation of coupling reversed phase, aqueous normal phase, and hydrophilic interaction liquid chromatography with Orbitrap mass spectrometry for metabolomic studies of human urine. Anal. Chem. 2012, 84, 1994–2001. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.K.; Ma, C.Y. Retention index scale for liquid-liquid chromatography. J. Chromatogr. A 1979, 169, 107–115. [Google Scholar] [CrossRef]
- Bogusz, M.; Aderjan, R. Corrected retention indices in HPLC: Their use for the identification of acidic and neutral drugs. J. Anal. Toxicol. 1988, 12, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.F.; Zhang, T.Y.; Qin, L.L.; Li, X.M.; Zheng, S.J.; Feng, Y.Q. Method to calculate the retention index in hydrophilic interaction liquid chromatography using normal fatty acid derivatives as calibrants. Anal. Chem. 2019, 91, 6057–6063. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Ventura, D.; Prince, J.T. LC-MS alignment in theory and practice: A comprehensive algorithmic review. Brief. Bioinf. 2015, 16, 104–117. [Google Scholar] [CrossRef]
- Tsugawa, H.; Nakabayashi, R.; Mori, T.; Yamada, Y.; Takahashi, M.; Rai, A.; Sugiyama, R.; Yamamoto, H.; Nakaya, T.; Yamazaki, M.; et al. A cheminformatics approach to characterize metabolomes in stable-isotope-labeled organisms. Nat. Methods 2019, 16, 295–298. [Google Scholar] [CrossRef]
- Rothwell, J.A.; Fillâtre, Y.; Martin, J.F.; Lyan, B.; Pujos-Guillot, E.; Fezeu, L.; Hercberg, S.; Comte, B.; Galan, P.; Touvier, M.; et al. New biomarkers of coffee consumption identified by the non-targeted metabolomic profiling of cohort study subjects. PLoS ONE 2014, 9, e93474. [Google Scholar] [CrossRef]
- Guo, X.; Bruins, A.P.; Covey, T.R. Characterization of typical chemical background interferences in atmospheric pressure ionization liquid chromatography-mass spectrometry. Rapid Commun. Mass Spectrom. 2006, 20, 3145–3150. [Google Scholar] [CrossRef]
- Keller, B.O.; Sui, J.; Young, A.B.; Whittal, R.M. Interferences and contaminants encountered in modern mass spectrometry. Anal. Chim. Acta 2008, 627, 71–81. [Google Scholar] [CrossRef]
- Tsugawa, H.; Hayakawa, E.; Miura, D. The guide for metabolite annotation/identification in untargeted metabolomics. J. Mass Spectrom. Soc. Jpn. 2017, 65, 203–209. [Google Scholar] [CrossRef]
- RIKEN PRIMe Spectra Download. Available online: http://prime.psc.riken.jp/Metabolomics_Software/MS-DIAL/index.html (accessed on 10 June 2019).
- MassBank of North America. Available online: http://mona.fiehnlab.ucdavis.edu/ (accessed on 10 June 2019).
- NIST 17 Libraries and Software. Available online: https://chemdata.nist.gov/dokuwiki/doku.php?id=chemdata:nist17 (accessed on 10 June 2019).
- Dührkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Böcker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [PubMed]
- Haug, K.; Salek, R.M.; Conesa, P.; Hastings, J.; de Matos, P.; Rijnbeek, M.; Mahendraker, T.; Williams, M.; Neumann, S.; Rocca-Serra, P.; et al. MetaboLights—An open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 2013, 41, D781–D786. [Google Scholar] [CrossRef] [PubMed]
- Stanstrup, J.; Neumann, S.; Vrhovšek, U. PredRet: Prediction of retention time by direct mapping between multiple chromatographic systems. Anal. Chem. 2015, 87, 9421–9428. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S.; et al. Metabolomics workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinf. 2010, 11, 395. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Kuhl, C.; Tautenhahn, R.; Bottcher, C.; Larson, T.R.; Neumann, S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef]
- Broeckling, C.D.; Afsar, F.A.; Neumann, S.; Ben-Hur, A.; Prenni, J.E. RAMClust: A novel feature clustering method enables spectral-matching-based annotation for metabolomics data. Anal. Chem. 2014, 86, 6812–6817. [Google Scholar] [CrossRef]
- Senan, O.; Aguilar-Mogas, A.; Navarro, M.; Capellades, J.; Noon, L.; Burks, D.; Yanes, O.; Guimera, R.; Sales-Pardo, M. CliqueMS: A computational tool for annotating in-source metabolite ions from LC-MS untargeted metabolomics data based on a coelution similarity network. Bioinformatics 2019, 35, 4089–4097. [Google Scholar] [CrossRef] [PubMed]
- mzCloud. Available online: https://www.mzcloud.org/ (accessed on 14 August 2019).
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag relaunched: Incorporating strategies beyond in silico fragmentation. J. Cheminf. 2016, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]
- MS-DIAL. Available online: http://prime.psc.riken.jp/Metabolomics_Software/MS-DIAL/index2.html (accessed on 14 August 2019).





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tada, I.; Tsugawa, H.; Meister, I.; Zhang, P.; Shu, R.; Katsumi, R.; Wheelock, C.E.; Arita, M.; Chaleckis, R. Creating a Reliable Mass Spectral–Retention Time Library for All Ion Fragmentation-Based Metabolomics. Metabolites 2019, 9, 251. https://doi.org/10.3390/metabo9110251
Tada I, Tsugawa H, Meister I, Zhang P, Shu R, Katsumi R, Wheelock CE, Arita M, Chaleckis R. Creating a Reliable Mass Spectral–Retention Time Library for All Ion Fragmentation-Based Metabolomics. Metabolites. 2019; 9(11):251. https://doi.org/10.3390/metabo9110251
Chicago/Turabian StyleTada, Ipputa, Hiroshi Tsugawa, Isabel Meister, Pei Zhang, Rie Shu, Riho Katsumi, Craig E. Wheelock, Masanori Arita, and Romanas Chaleckis. 2019. "Creating a Reliable Mass Spectral–Retention Time Library for All Ion Fragmentation-Based Metabolomics" Metabolites 9, no. 11: 251. https://doi.org/10.3390/metabo9110251
APA StyleTada, I., Tsugawa, H., Meister, I., Zhang, P., Shu, R., Katsumi, R., Wheelock, C. E., Arita, M., & Chaleckis, R. (2019). Creating a Reliable Mass Spectral–Retention Time Library for All Ion Fragmentation-Based Metabolomics. Metabolites, 9(11), 251. https://doi.org/10.3390/metabo9110251