Metabolomic Profiling and Cytotoxic Tetrahydrofurofuran Lignans Investigations from Premna odorata Blanco

 ,
, 
 , ,
, , 
Abstract
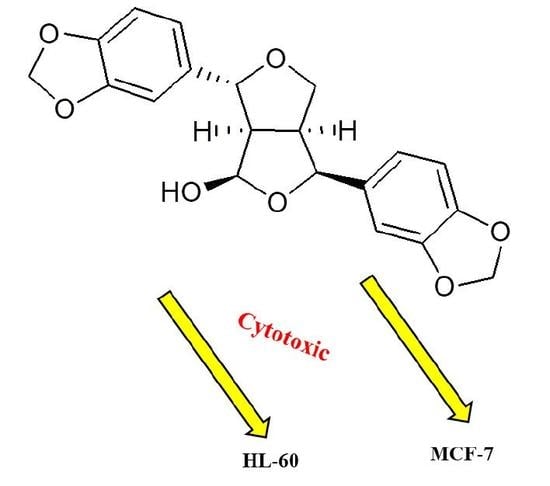
:
1. Introduction
2. Results and Discussion
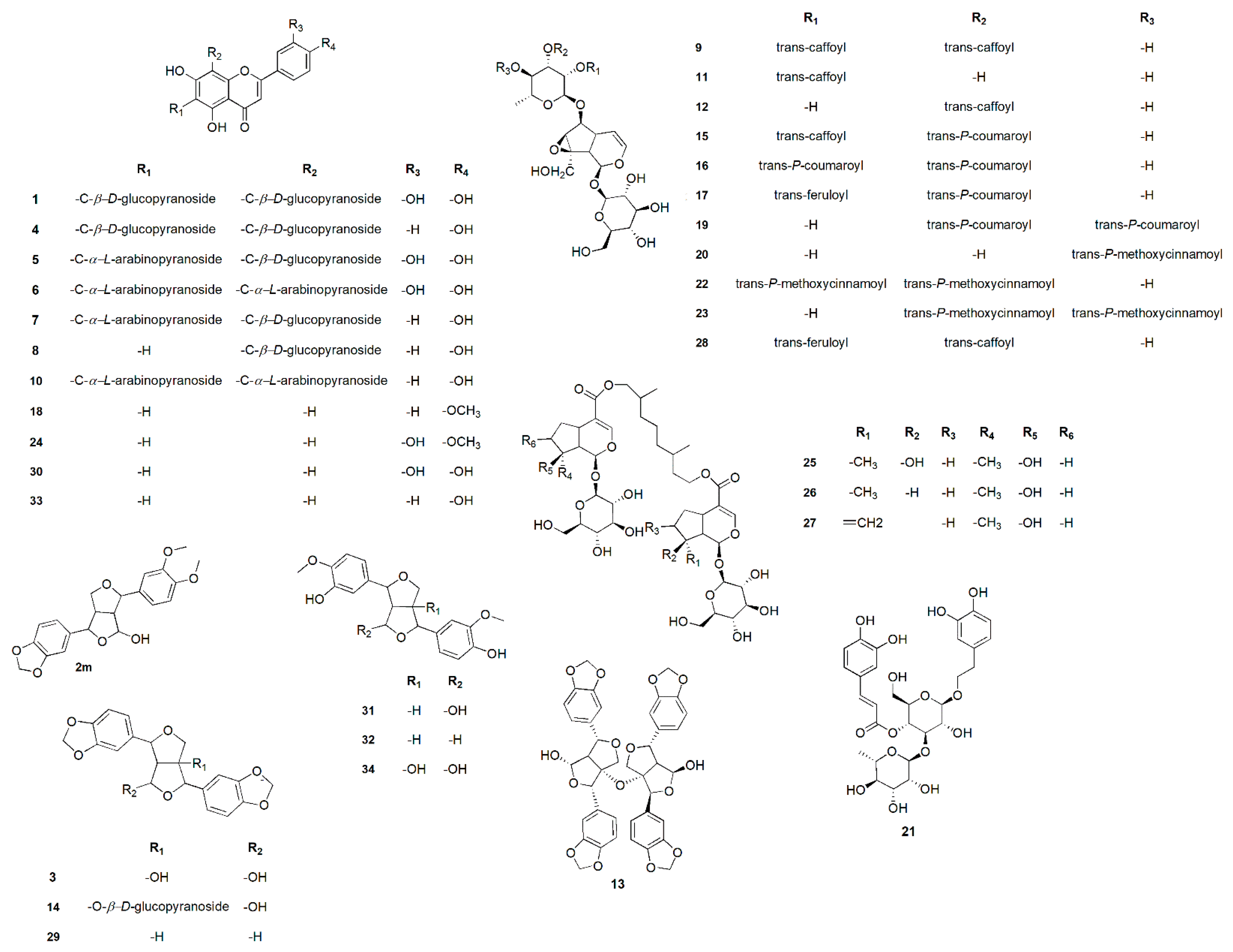
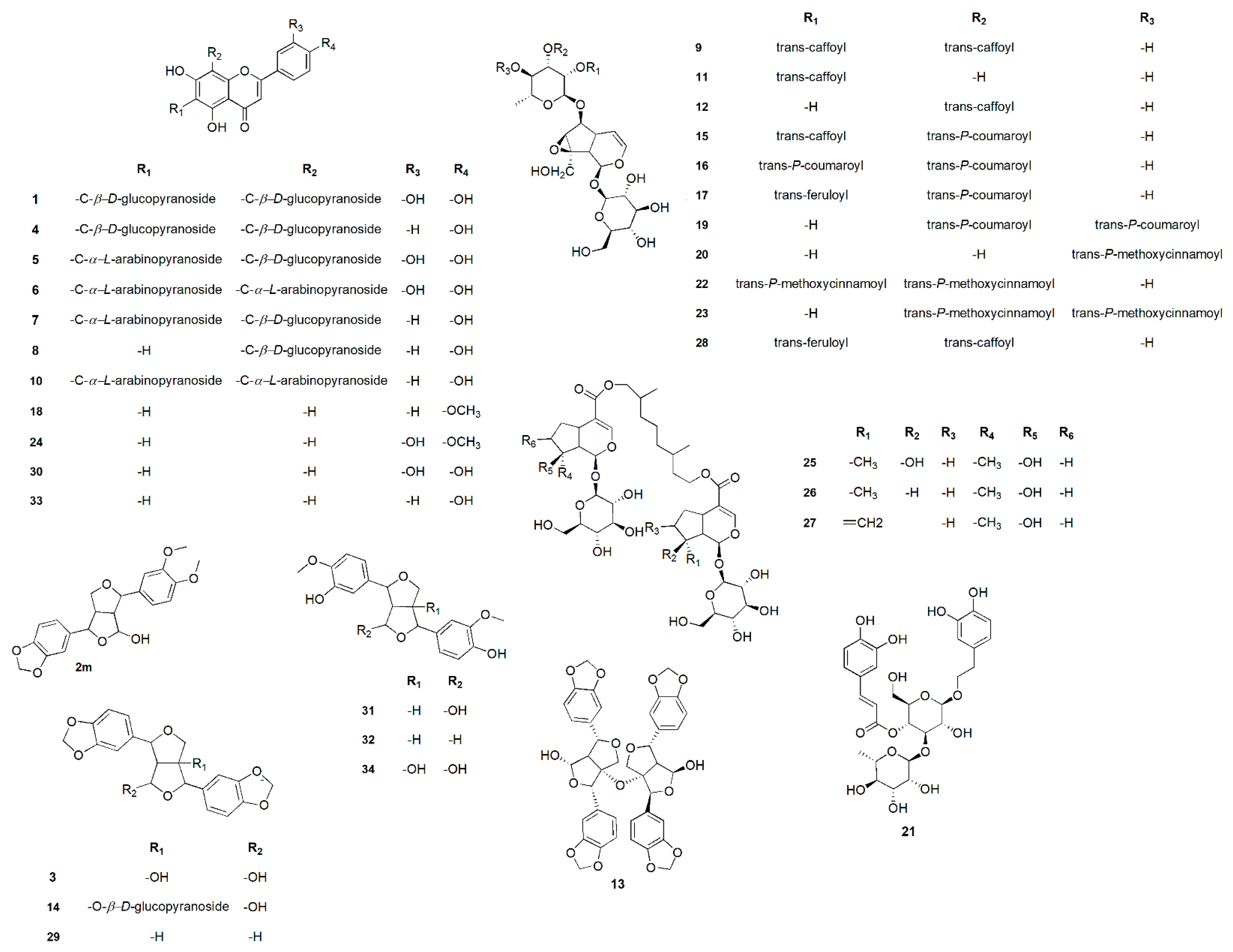
2.1. Metabolomic Analysis
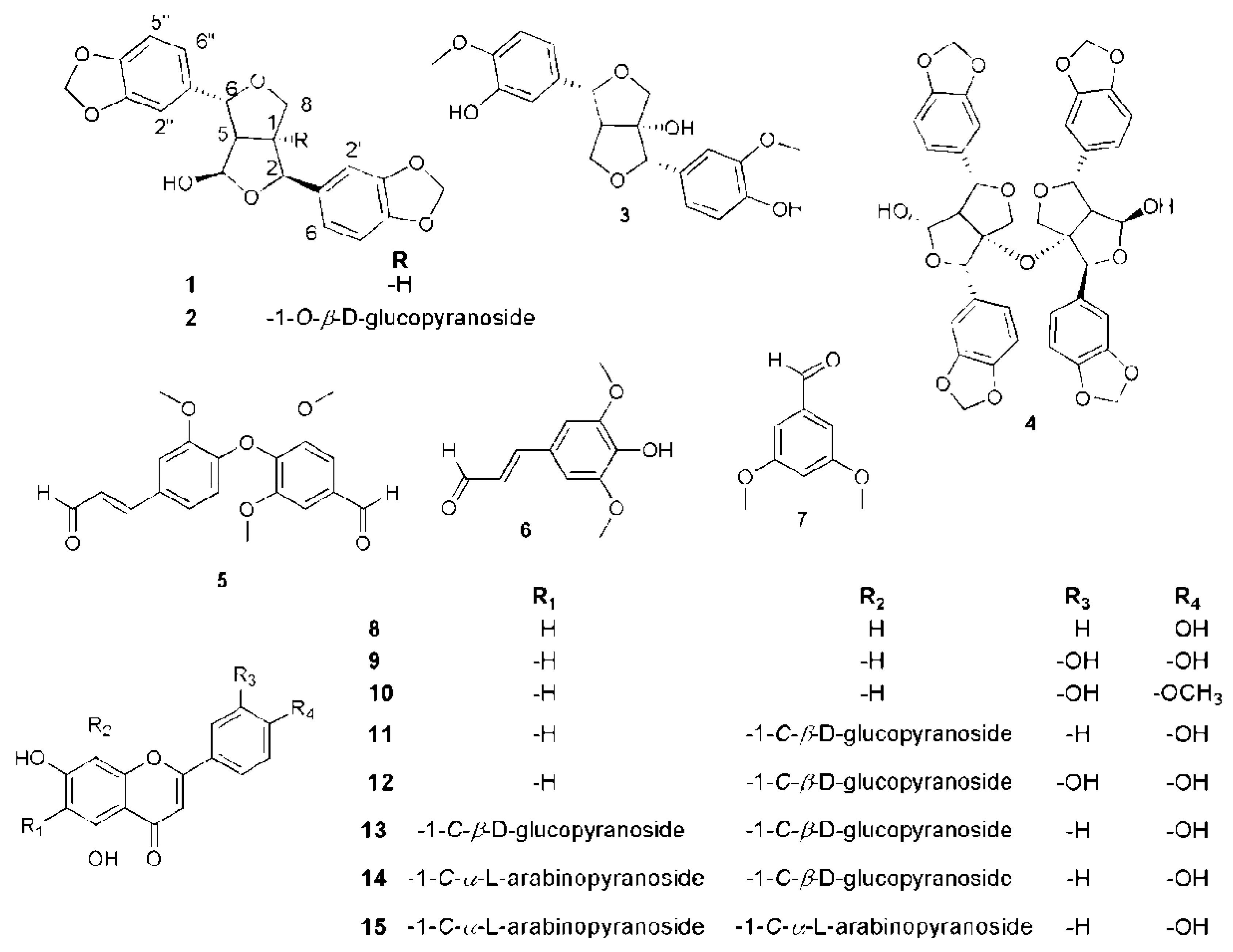
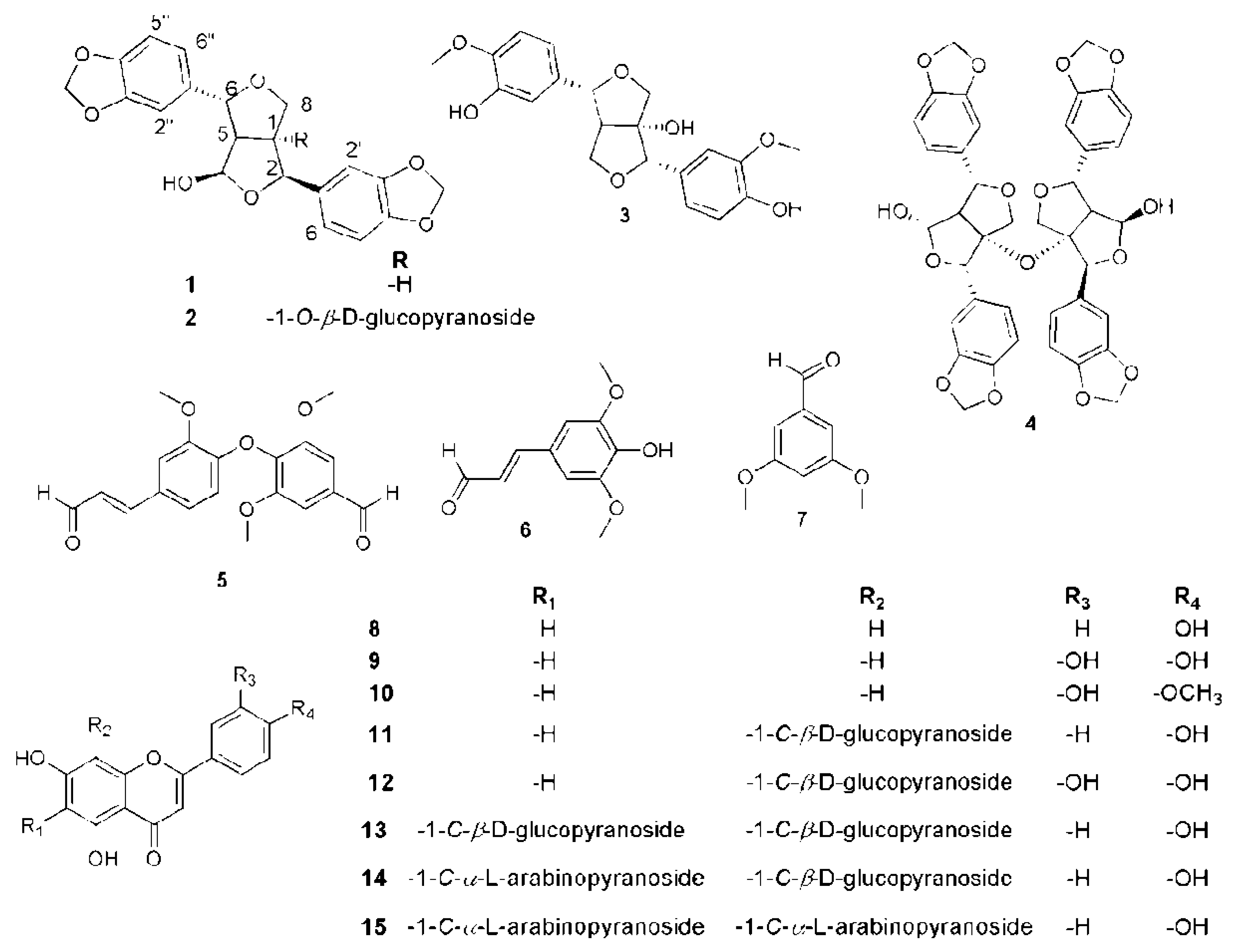
2.2. Phytochemical Investigation of P. odorata Bark
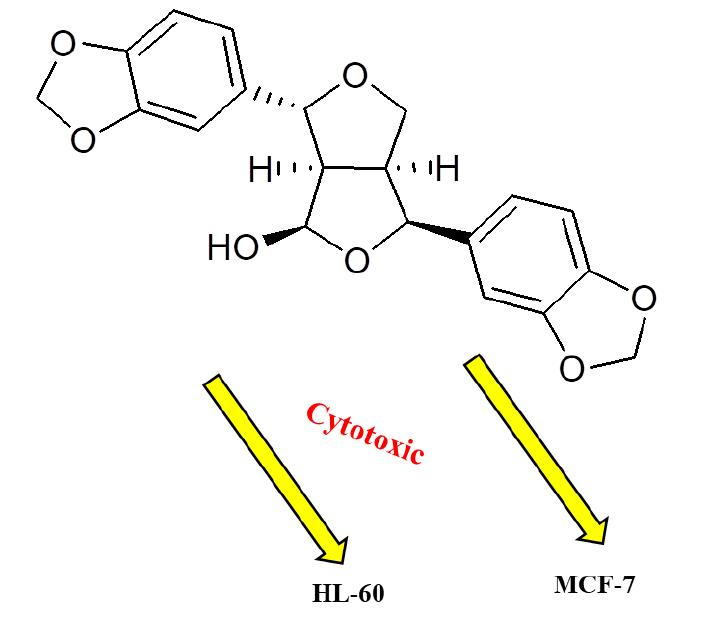
2.3. Cytotoxic Activity
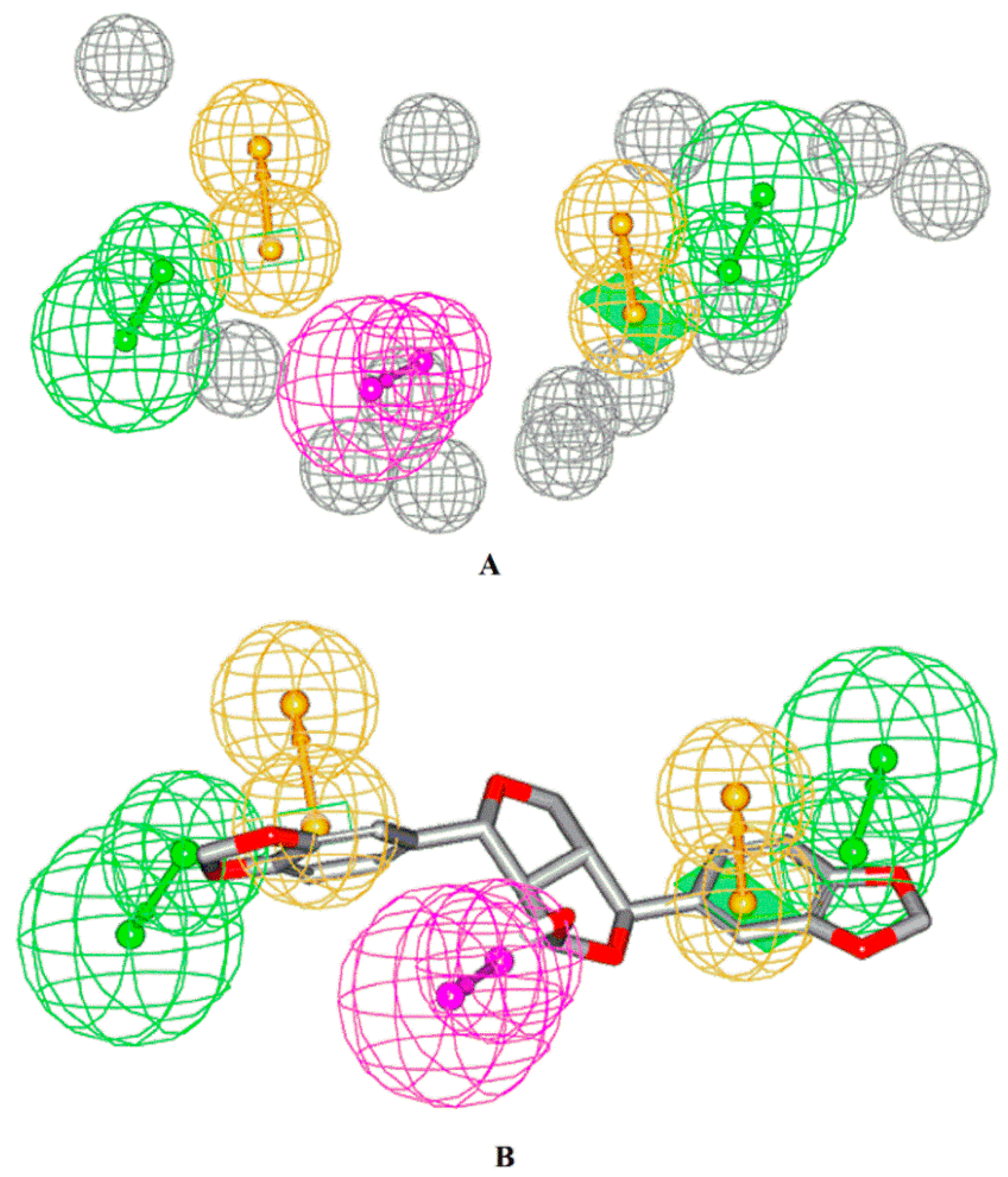
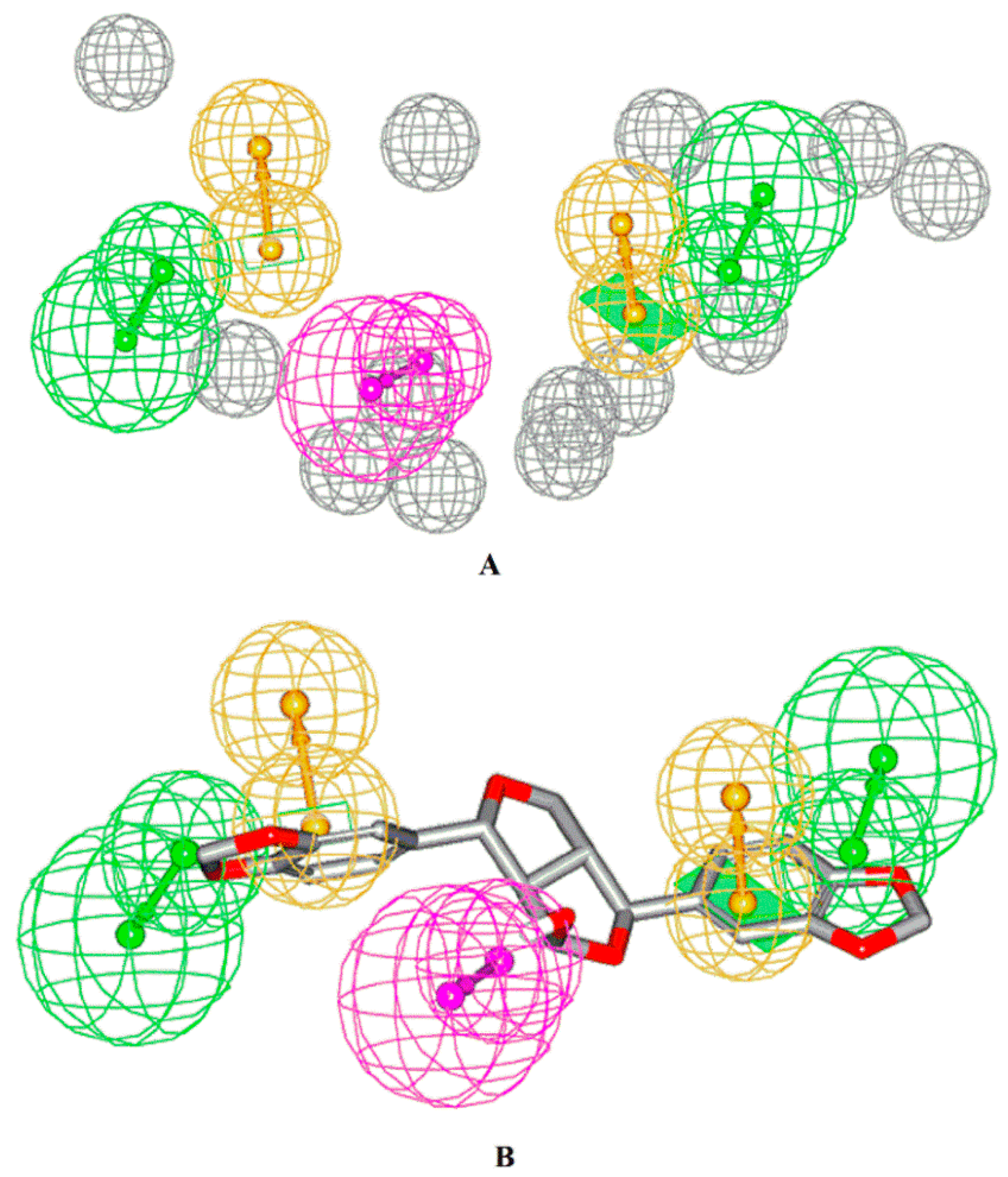
2.4. Pharmacophore Model Generation
3. Materials and Methods
3.1. Plant Material
3.2. Chemicals and Reagents
3.3. Apparatus
3.4. Extraction and Fractionation of Plant Material
3.5. Metabolomic Analysis Procedure
3.6. Isolation and Purification of Compounds
3.7. Cell Cultures
3.8. Cytotoxic Assessment
3.9. Pharmacophore Model Generation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, K.H.; Moon, E.; Kim, S.Y.; Choi, S.U.; Lee, K.R. Lignan constituents of Tilia amurensis and their biological evaluation on antitumor and anti-inflammatory activities. Food Chem. Toxicol. 2012, 50, 3680–3686. [Google Scholar] [CrossRef] [PubMed]
- Olano, C.; Méndez, C.; Salas, J.A. Antitumor compounds from actinomycetes: From gene clusters to new derivatives by combinatorial biosynthesis. Nat. Prod. Rep. 2009, 26, 628–660. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Lirio, S.B.; Macabeo, A.P.G.; Paragas, E.M.; Knorn, M.; Kohls, P.; Franzblau, S.G.; Wang, Y.; Aguinaldo, M.A.M. Antitubercular constituents from Premna odorata Blanco. J. Ethnopharmacol. 2014, 154, 471–474. [Google Scholar] [CrossRef]
- Elmaidomy, A.H.; Mohyeldin, M.M.; Ibrahim, M.M.; Hassan, H.M.; Amin, E.; Rateb, M.E.; Hetta, M.H.; El Sayed, K.A. Acylated iridoids and rhamnopyranoses from Premna odorata (lamiaceae) as novel mesenchymal epithelial transition factor receptor inhibitors for the control of breast cancer. Phytother. Res. 2017, 31, 1546–1556. [Google Scholar] [CrossRef]
- Elmaidomy, A.H.; Hassan, H.M.; Amin, E.; Mohamed, W.; Hetta, M.H. Premna odorata volatile oil as a new mycobacterium tuberculosis growth inhibitor for the control of tuberculosis disease. Eur. J. Med. Plants 2017, 21, 1–11. [Google Scholar] [CrossRef]
- Otsuka, H.; Kashima, N.; Hayashi, T.; Kubo, N.; Yamasaki, K.; Padolina, W.G. Premnaodorosides A, B and C, iridoid glucoside diesters of an acyclic monoterpenediol from leaves of Premna odorata. Phytochemistry 1992, 31, 3129–3133. [Google Scholar] [CrossRef]
- Otsuka, H.; Kubo, N.; Sasaki, Y.; Yamasaki, K.; Takeda, Y.; Seki, T. Iridoid diglycoside monoacyl esters from stems of Premna japonica. Phytochemistry 1991, 30, 1917–1920. [Google Scholar] [CrossRef]
- Otsuka, H.; Kubo, N.; Yamasaki, K.; Padolina, W.G. Premnosides A–D: Diacyl 6-O-α-l-rhamnopyranosylcatalpols from Premna odorata. Phytochemistry 1989, 28, 3063–3067. [Google Scholar] [CrossRef]
- Rambla, J.L.; Lopez, M.P.; Belles, J.M.; Granell, A. Metabolomic Profiling of Plant Tissues, in Plant Functional Genomics; Springer: New York, NY, USA, 2015; pp. 221–235. [Google Scholar]
- Clarke, C.J.; Haselden, J.N. Metabolic profiling as a tool for understanding mechanisms of toxicity. Toxicol. Pathol. 2008, 36, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Allwood, J.W.; Goodacre, R. An introduction to liquid chromatography–mass spectrometry instrumentation applied in plant metabolomic analyses. Phytochem. Anal. 2010, 21, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Veitch, N.C.; Houghton, P.J.; Simmonds, M.S. Flavone C-Glycosides from Viola yedoensis M Akino. Chem. Pharm. Bull. 2003, 51, 1204–1207. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.H.; Jeong, J.E.; Choi, E.J.; Moon, Y.H.; Woo, E.R. A new furofuran lignan from Geranium thunbergii Sieb. et Zucc. Arch. Pharmacal. Res. 2006, 29, 1109. [Google Scholar] [CrossRef] [PubMed]
- Anjaneyulu, A.S.; Rao, K.J.; Rao, V.K.; Row, L.R.; Subrahmanyam, C.; Pelter, A.; Ward, R.S. The structures of lignans from Gmelina arborea Linn. Tetrahedron 1975, 31, 1277–1285. [Google Scholar] [CrossRef]
- Desta, Z.Y.; Sewald, N.; Majinda, R.R. New flavonoids from the stem bark of Erythrina caffra Thunb. Nat. Prod. Res. 2014, 28, 667–673. [Google Scholar] [CrossRef]
- Markham, K.R.; Mues, R.; Stoll, M.; Zinsmeister, H.D. NMR spectra of flavone di-C-glycosides from Apometzgeria pubescens and the detection of rotational isomerism in 8-C-hexosylflavones. Z. Nat. C 1987, 42, 1039–1042. [Google Scholar] [CrossRef]
- Yadav, D.; Masood, N.; Luqman, S.; Brindha, P.; Gupta, M.M. Antioxidant furofuran lignans from Premna integrifolia. Ind. Crops Prod. 2013, 41, 397–402. [Google Scholar] [CrossRef]
- Ju, Y.; Still, C.C.; Sacalis, J.N.; Li, J.; Ho, C.T. Cytotoxic coumarins and lignans from extracts of the northern prickly ash (Zanthoxylum americanum). Phytotherp. Res. 2001, 15, 441–443. [Google Scholar] [CrossRef]
- Abe, F.; Yamauchi, T. 9α-Hydroxypinoresinol, 9α-hydroxymedioresinol and related lignans from Allamanda neriifolia. Phytochemistry 1988, 27, 575–577. [Google Scholar] [CrossRef]
- Casabuono, A.C.; Pomillo, A.B. Lignans and a stilbene from Festuca argentina. Phytochemistry 1994, 35, 479–483. [Google Scholar] [CrossRef]
- Benabdelaziz, I.; Haba, H.; Lavaud, C.; Harakat, D.; Benkhaled, M. Lignans and Other Constituents from Helianthemum sessiliflorum Pers. Rec. Nat. Prod. 2015, 9, 342. [Google Scholar]
- Li, N.; Lee, H.S.; Zhang, N.; Sun, Y.N.; Li, J.L.; Xing, S.S.; Chen, J.G.; Cui, L. Two new diphenyl ethers from Acanthopanax senticosus (Rupr. & Maxim.) Harms with PTP1B inhibitory activity. Phytochem. Lett. 2015, 13, 286–289. [Google Scholar]
- Zhang, X.; Guo, P.; Sun, G.; Chen, S.; Yang, M.; Fu, N.; Wu, H.; Xu, X. Phenolic compounds and flavonoids from the fruits of Pandanus tectorius Soland. J. Med. Plants Res. 2012, 6, 2622–2626. [Google Scholar]
- Elmasry, S.; Hammoda, H.M.; Radwan, M.M.; Ross, S.A.; Zaatout, H.H. C-flavonoidal glycosides from Erythrina caffra flowers. Nat. Prod. Sci. 2010, 16, 217–222. [Google Scholar]
- Sok, D.E.; Cui, H.S.; Kim, M.R. Isolation and boactivities of furfuran type lignan compounds from edible plants. Recent Pat. Food Nutr. Agric. 2009, 1, 87–95. [Google Scholar] [CrossRef]
- Ren, Y.; Yuan, C.; Deng, Y.; Kanagasabai, R.; Ninh, T.N.; Tu, V.T.; Chai, H.B.; Soejarto, D.D.; Fuchs, J.R.; Yalowich, J.C. Cytotoxic and natural killer cell stimulatory constituents of Phyllanthus songboiensis. Phytochemistry 2015, 111, 132–140. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, J.; Yu, Y.; Kim, Y.C. Inhibition of phospholipase Cγ1 and cancer cell proliferation by lignans and flavans from Machilus thunbergii. Arch. Pharmacal. Res. 2004, 27, 1043–1047. [Google Scholar] [CrossRef]
- Khanfar, M.A.; AbuKhader, M.M.; Alqtaishat, S.; Taha, M.O. Pharmacophore modeling, homology modeling, and in silico screening reveal mammalian target of rapamycin inhibitory activities for sotalol, glyburide, metipranolol, sulfamethizole, glipizide, and pioglitazone. J. Mol. Graph. Model 2013, 42, 39–49. [Google Scholar] [CrossRef]
- Khanfar, M.A.; Taha, M.O. Elaborate ligand-based modeling coupled with multiple linear regression and k nearest neighbor QSAR analyses unveiled new nanomolar mTOR inhibitors. J. Chem. Inf. Model 2013, 53, 2587–2612. [Google Scholar] [CrossRef]
- Stahl, E. Thin Layer Chromatography, 2nd ed.; Springer Verlage: Berlin/Heidelberg, Germany; New York, NY, USA, 1970; p. 810. [Google Scholar]
- Markham, K.R. Techniques of Flavonoids Identification; Academic Press: London, UK, 1982. [Google Scholar]
- Abdelmohsen, U.R.; Cheng, C.; Viegelmann, C.; Zhang, T.; Grkovic, T.; Ahmed, S.; Quinn, R.J.; Hentschel, U.; Edrada-Ebel, R. Dereplication strategies for targeted isolation of new antitrypanosomal actinosporins A and B from a marine sponge associated-Actinokineospora sp. EG49. Mar. Drugs 2014, 12, 1220–1244. [Google Scholar] [CrossRef]
- Mojarrab, M.; Lagzian, M.S.; Emami, S.A.; Asili, J.; Tayarani-Najaran, Z. In vitro anti-proliferative and apoptotic activity of different fractions of Artemisia armeniaca. Rev. Bras. Farmacogn. 2013, 23, 783–788. [Google Scholar] [CrossRef]
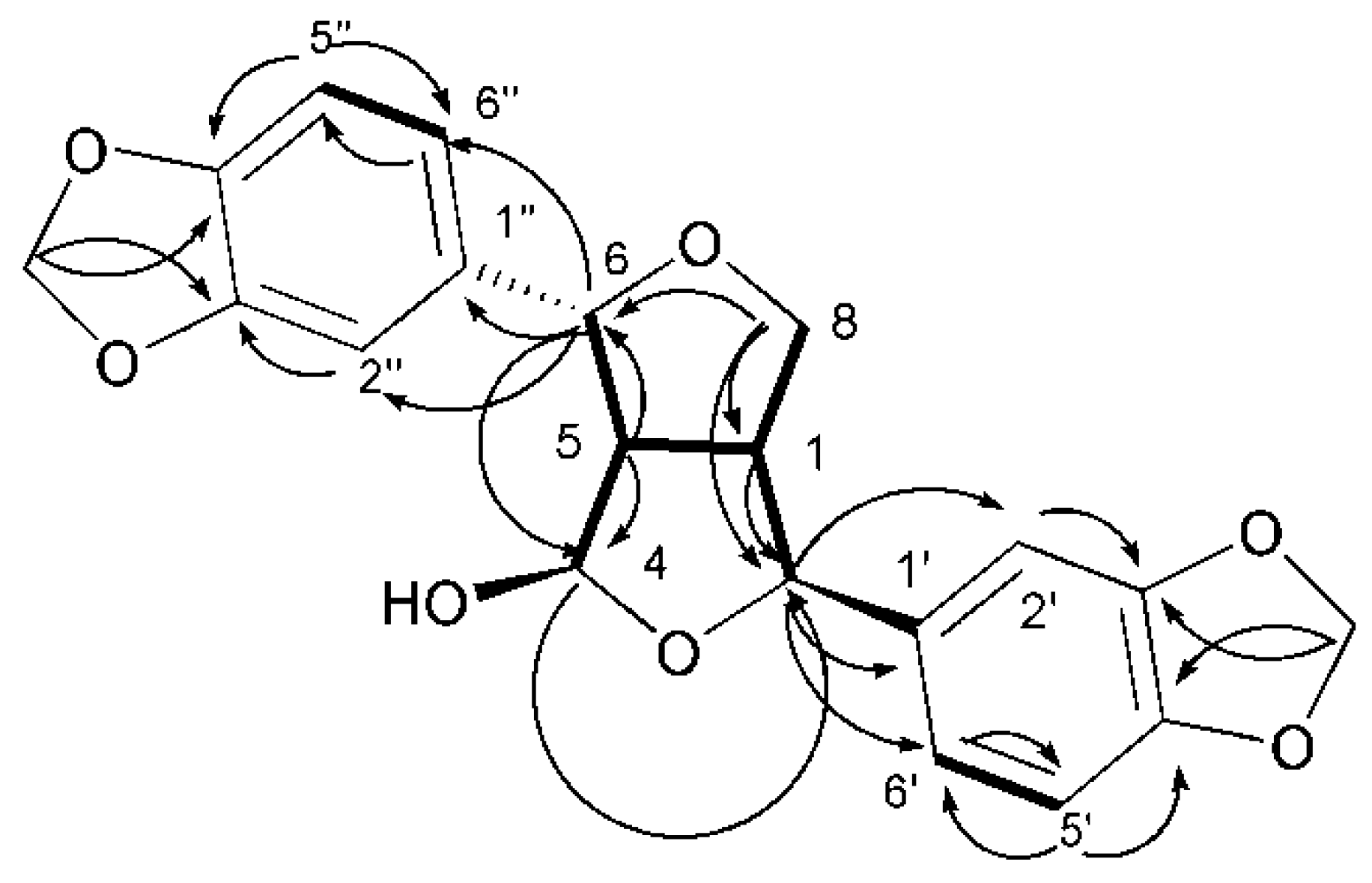
 ) and 1H–1H COSY (
) and 1H–1H COSY (  ) correlations of compound 35.
) correlations of compound 35.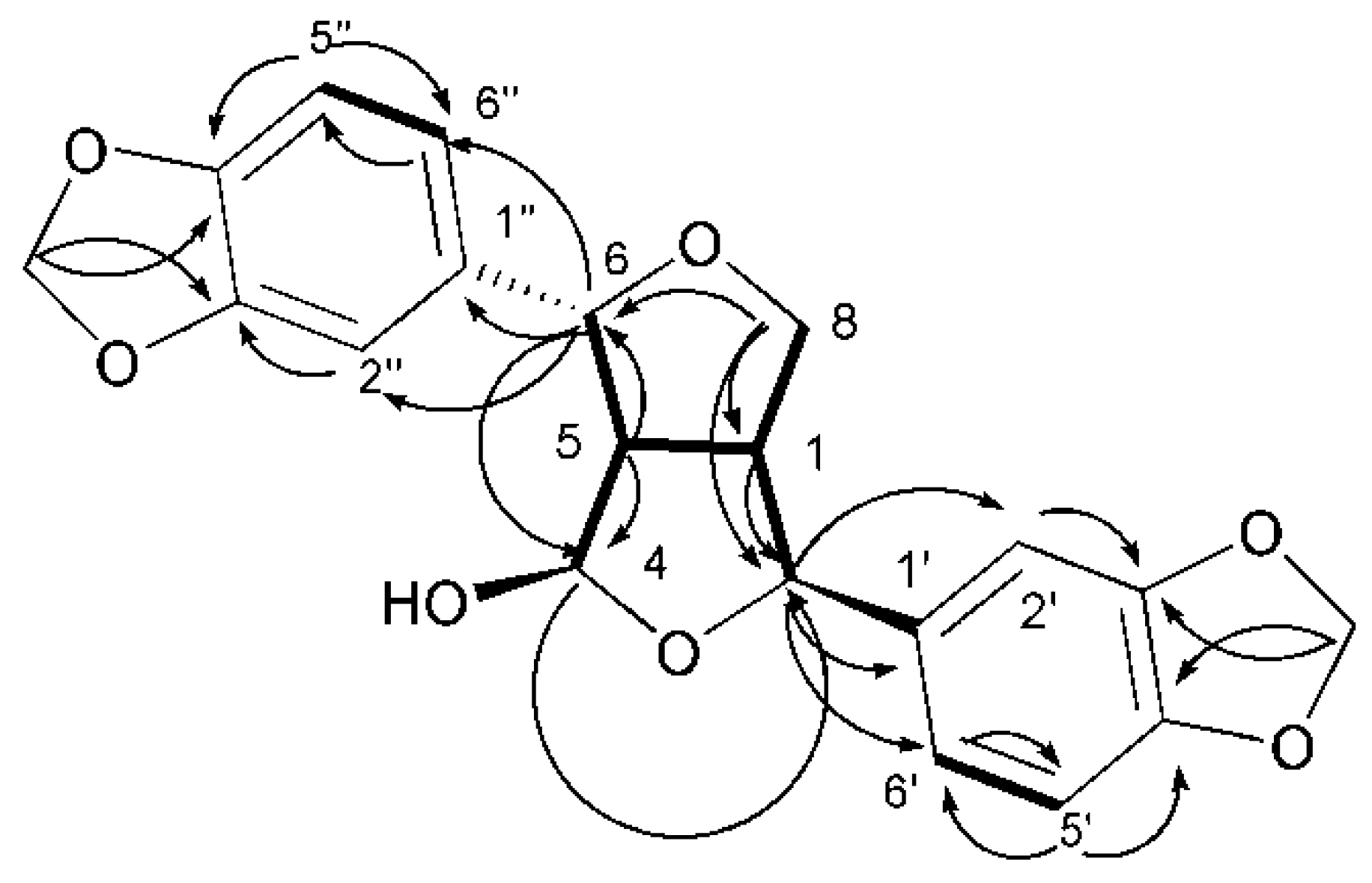

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Metabolites Name | Original Source | MF | RT(min.) | m/z | B. | W. | Y. | Fl. | Fr. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Luteolin-6,8-di-c-β-d-glucopyranoside | Viola yedoensis | C27H30O16 | 7.54 | 611.1915 | + | + | |||
| 2 | 9-hydroxy-3’,4’-dimethoxy-3,4-methylenedioxy-7,9’:7’,9-diepoxylignan | Geranium thunbergii | C21H22O7 | 7.60 | 387.1276 | + | + | |||
| 3 | 4,8-dihydroxy sesamin | Gmelina arborea | C20H18O8 | 7.71 | 387.1323 | + | + | |||
| 4 | Vicenin-2 | Erythrina caffra | C27H30O15 | 8.25 | 595.2334 | + | + | |||
| 5 | luteolin 6-c-β-d-glucopyranoside, 8-c-α-l-arabinopyranoside | Apometzgeria pubescens | C26H28O15 | 8.65 | 581.1560 | + | + | |||
| 6 | Luteolin-6,8-di-c-α-l-arabinopyranoside | Apometzgeria pubescens | C25H26O14 | 8.89 | 551.1592 | + | + | |||
| 7 | Schaftoside-2 | Apometzgeria pubescens | C26H28O14 | 9.11 | 565.2288 | + | + | |||
| 8 | Vitexin | Premna odorata | C21H20O10 | 9.71 | 433.1361 | + | + | + | + | + |
| 9 | Premnoside A | Premna odorata | C39H44O20 | 11.37 | 833.2746 | + | + | |||
| 10 | Apigenin-6,8-di-c-α-l-arabinopyranoside | Apometzgeria pubescens | C25H26O13 | 11.81 | 535.2889 | + | + | |||
| 11 | 6- o-α- l-(2″-o-trans-caffoyl) rhamnopyranosylcatalpol | Premna odorata | C30H38O17 | 11.98 | 671.1910 | + | + | |||
| 12 | 6- o-α- l-(3″-o-trans-caffoyl) rhamnopyranosylcatalpol | Premna odorata | C30H38O17 | 12.03 | 671.1910 | + | + | |||
| 13 | Premnadimer A | Premna integrifolia | C40H34O15 | 12.22 | 793.3148 | + | + | |||
| 14 | 4β-hydroxyasarinin-1-o-β-d-glucopyranoside | Premna integrifolia | C26H28O13 | 12.53 | 549.1555 | + | + | |||
| 15 | Premnoside B | Premna odorata | C39H44O19 | 12.61 | 817.2282 | + | + | |||
| 16 | Premnoside F | Premna odorata | C39H44O18 | 13.00 | 801.2404 | + | + | + | ||
| 17 | Premnoside D | Premna odorata | C40H46O19 | 13.09 | 831.2411 | + | + | |||
| 18 | Acacetin | Premna odorata | C16H12O5 | 13.60 | 285.1126 | + | + | + | + | + |
| 19 | Premnoside E | Premna odorata | C39H44O18 | 13.72 | 801.2404 | + | + | + | ||
| 20 | 6- o-α- l-(4’’-o-trans-p-methoxycinnamoyl) rhamnopyranosylcatalpol | Premna odorata | C31H40O16 | 14.10 | 669.1634 | + | + | |||
| 21 | Verbascoside | Premna odorata | C29H36O15 | 14.47 | 625.1396 | + | + | + | + | + |
| 22 | Premnoside H | Premna odorata | C41H48O18 | 14.54 | 829.2011 | + | + | + | ||
| 23 | Premnoside G | Premna odorata | C41H48O18 | 14.73 | 829.2011 | + | + | + | ||
| 24 | Diosmetin | Premna odorata | C16H12O6 | 15.09 | 301.2947 | + | + | + | + | + |
| 25 | Premnaodoroside A | Premna odorata | C42H66O20 | 15.94 | 891.3561 | + | + | |||
| 26 | Premnaodoroside B | Premna odorata | C42H66O19 | 16.06 | 875.2437 | + | + | |||
| 27 | Premnaodoroside C | Premna odorata | C42H64O19 | 16.12 | 873.3192 | + | + | |||
| 28 | Premnoside C | Premna odorata | C40H46O20 | 16.21 | 847.2782 | + | + | |||
| 29 | Sesamin | Zanthoxylum senegalense | C20H18O6 | 18.21 | 355.1937 | + | + | |||
| 30 | Luteolin | Premna odorata | C15H10O6 | 18.66 | 309.2349 | + | + | + | + | + |
| 31 | 9-hydroxypinoresinol | Allamanda neriifolia | C20H22O7 | 19.07 | 379.2381 | + | + | |||
| 32 | Pinoresinol | Festuca argentina | C20H22O6 | 20.61 | 359.4261 | + | + | |||
| 33 | Apigenin | Premna odorata | C15H10O5 | 20.82 | 293.2147 | + | + | + | + | + |
| 34 | 1,5α-dihydroxypinoresinol | Helianthemum sessiliflorum | C20H22O8 | 22.25 | 391.2497 | + | + |
| Position | δC | δH (J in Hz) |
|---|---|---|
| 1 | 53.2,CH | 3.91 (dddd, 6.1, 7.1, 7.3, 11.1) |
| 2 | 83.3,CH | 4.84 (d,7.1) |
| 3 | ||
| 4 | 101.7,CH | 5.57 (s) |
| 5 | 62.1,CH | 2.93 (dd, 7.3, 7.5) |
| 6 | 88.1,CH | 4.96 (d, 10.1) |
| 7 | ||
| 8 | 72.2,CH2 | 4.01 (dd, 9.1, 11.1), 4.22 (dd,9.1, 6.1) |
| 1’ | 135.4,C | |
| 2’ | 107.1,CH | 7.06 (d,2.1) |
| 3’ | 147.3,C | |
| 4’ | 147.2,C | |
| 5’ | 108.1,CH | 6.81 (d,8.0) |
| 6’ | 119.2,CH | 6.80 (dd,2.1,8.0) |
| OCH2 | 101.4 | 5.41 (s) |
| 1’’ | 136.1,C | |
| 2’’ | 106.3,CH | 6.86 (d,2.0) |
| 3’’ | 147.4,C | |
| 4’’ | 147.3,C | |
| 5’’ | 108.2,CH | 6.89 (d,8.1) |
| 6’’ | 120.0,CH | 6.78 (dd,2.0,8.1) |
| OCH2 | 101.4 | 5.41 (s) |
| Compound | IC50 (µg/mL) a | ||
|---|---|---|---|
| HL-60 | MCF-7 | HT-29 | |
| 35 | 2.7 * | 4.2 * | NA |
| 36 | NA | NA | NA |
| 37 | NA | NA | NA |
| 38 | NA | NA | NA |
| Doxirubicin b | 0.08 | 0.68 | 501 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elmaidomy, A.H.; Mohammed, R.; M. Hassan, H.; I. Owis, A.; E. Rateb, M.; A. Khanfar, M.; Krischke, M.; J. Mueller, M.; Ramadan Abdelmohsen, U. Metabolomic Profiling and Cytotoxic Tetrahydrofurofuran Lignans Investigations from Premna odorata Blanco. Metabolites 2019, 9, 223. https://doi.org/10.3390/metabo9100223
Elmaidomy AH, Mohammed R, M. Hassan H, I. Owis A, E. Rateb M, A. Khanfar M, Krischke M, J. Mueller M, Ramadan Abdelmohsen U. Metabolomic Profiling and Cytotoxic Tetrahydrofurofuran Lignans Investigations from Premna odorata Blanco. Metabolites. 2019; 9(10):223. https://doi.org/10.3390/metabo9100223
Chicago/Turabian StyleElmaidomy, Abeer H., Rabab Mohammed, Hossam M. Hassan, Asmaa I. Owis, Mostafa E. Rateb, Mohammad A. Khanfar, Markus Krischke, Martin J. Mueller, and Usama Ramadan Abdelmohsen. 2019. "Metabolomic Profiling and Cytotoxic Tetrahydrofurofuran Lignans Investigations from Premna odorata Blanco" Metabolites 9, no. 10: 223. https://doi.org/10.3390/metabo9100223
APA StyleElmaidomy, A. H., Mohammed, R., M. Hassan, H., I. Owis, A., E. Rateb, M., A. Khanfar, M., Krischke, M., J. Mueller, M., & Ramadan Abdelmohsen, U. (2019). Metabolomic Profiling and Cytotoxic Tetrahydrofurofuran Lignans Investigations from Premna odorata Blanco. Metabolites, 9(10), 223. https://doi.org/10.3390/metabo9100223