Random Forest Analysis of Untargeted Metabolomics Data Suggests Increased Use of Omega Fatty Acid Oxidation Pathway in Drosophila Melanogaster Larvae Fed a Medium Chain Fatty Acid Rich High-Fat Diet


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. LC/MS and GC/MS Analysis
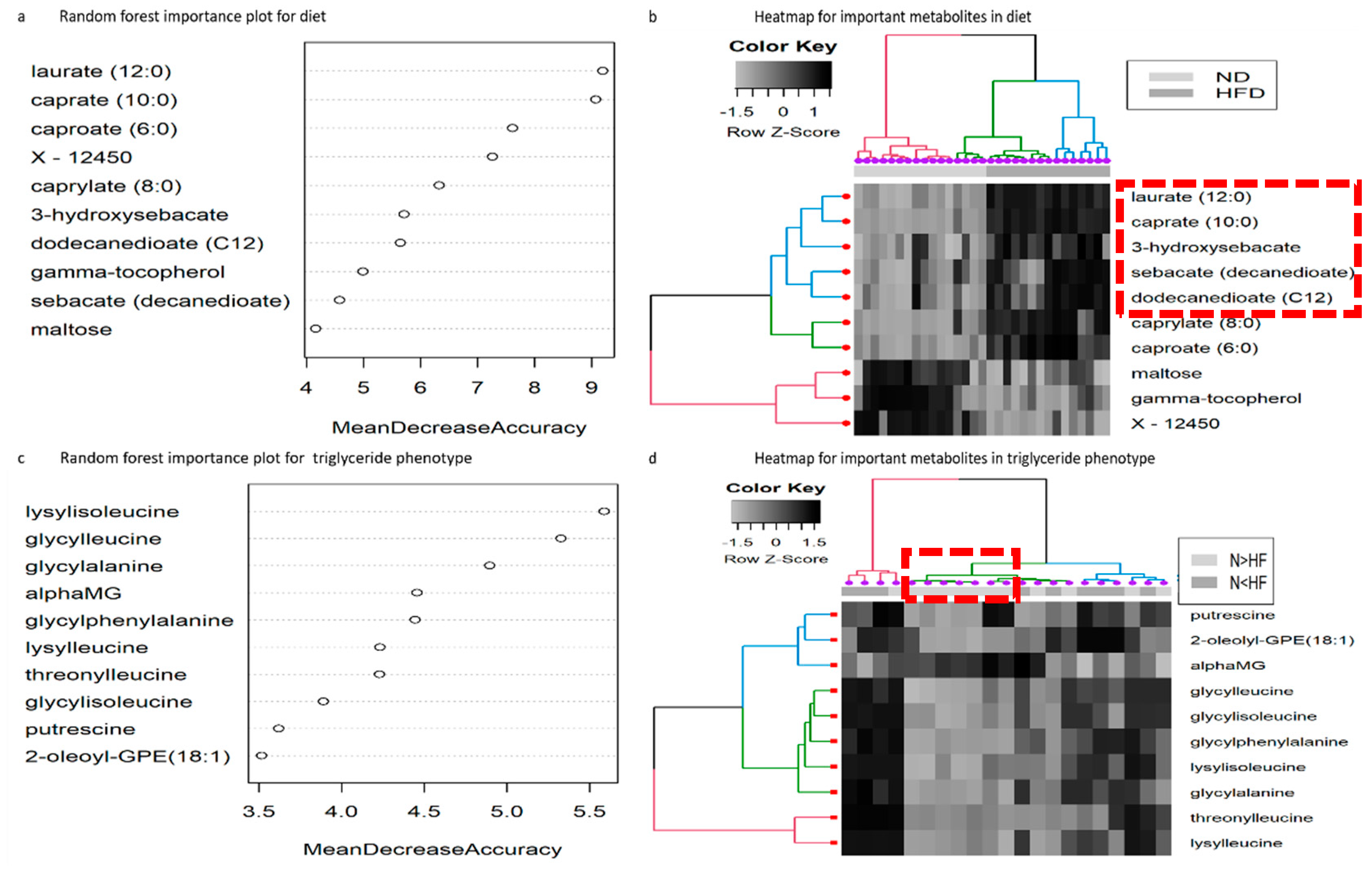
2.2. Random Forest Analysis for Diet
2.3. Random Forest Analysis for Reaction Norm Phenotypes
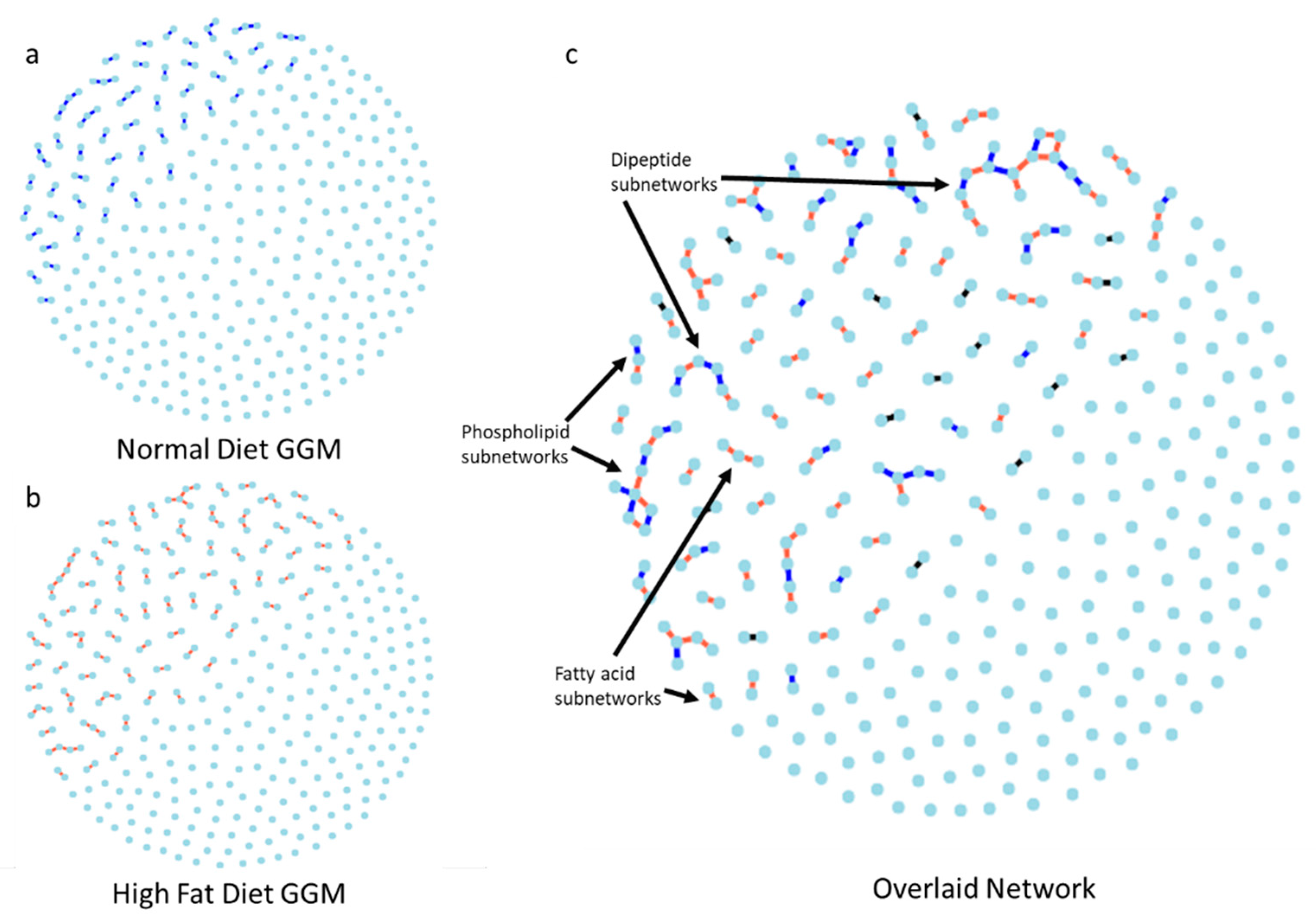
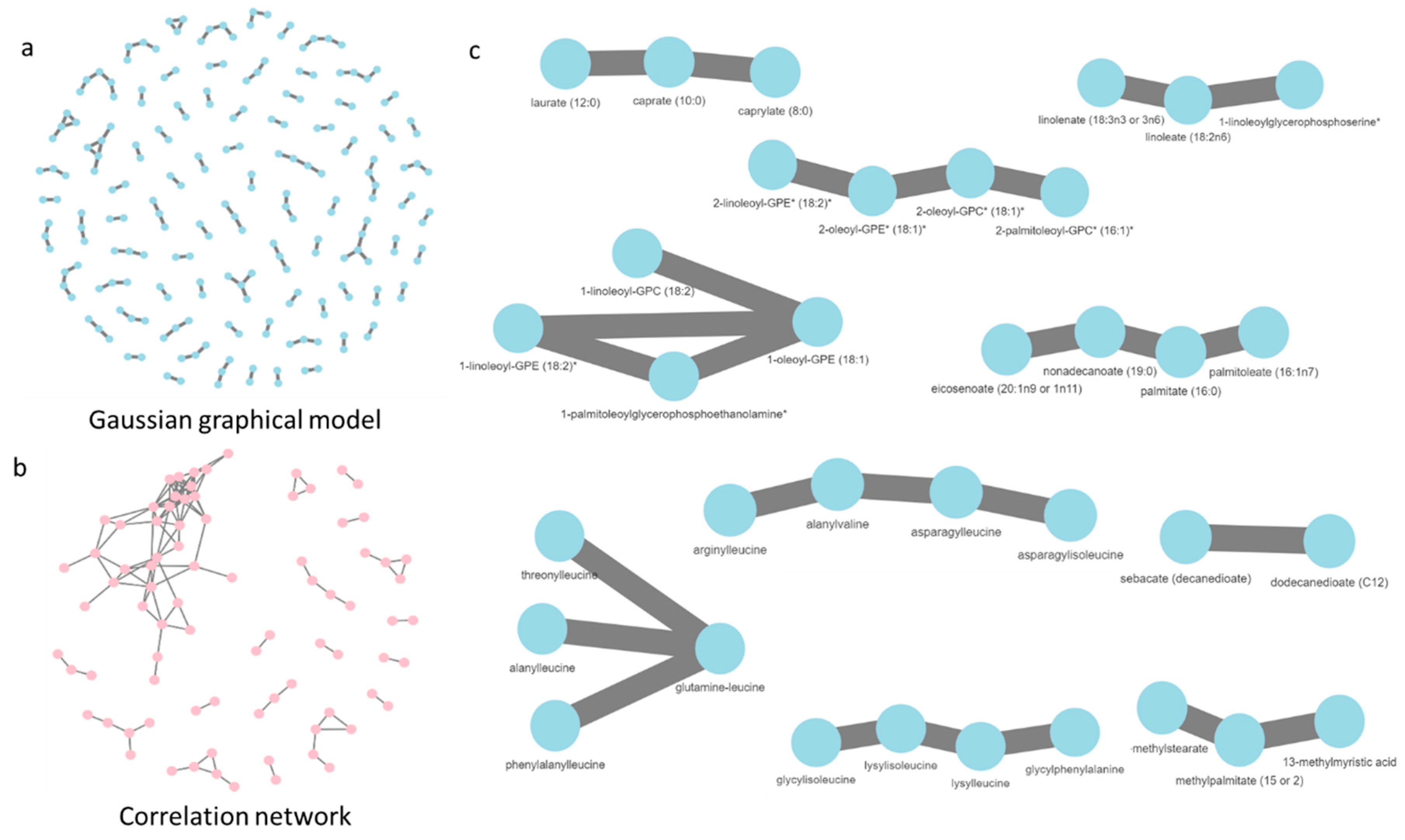
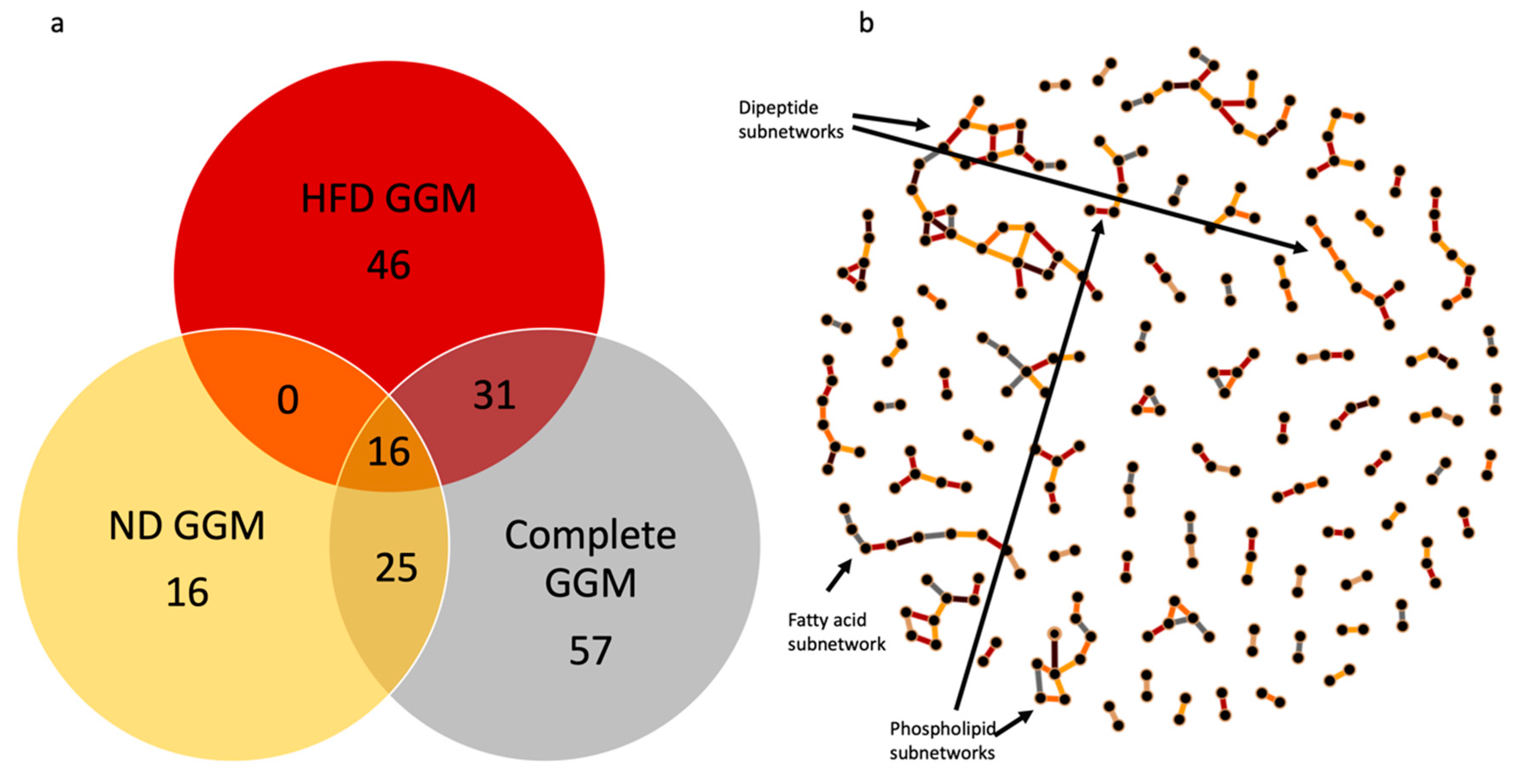
2.4. Gaussian Graphical Model of the Metabolic Pathways in Diet
2.5. The Role of Unknown Metabolites
3. Discussion
3.1. Effect of Diet on Lipid and Central Energy Metabolism
3.2. Effect of Triglyceride Storage on Lipid and Central Energy Metabolism
3.3. Network Analysis of Metabolites in ND and HFD Larvae
4. Materials and Methods
4.1. Sample Preparation
4.2. Liquid Chromatography/Mass Spectrometry (LC/MS, LC/MS2) Analysis
4.3. Gas Chromatography/Mass Spectrometry (GC/MS) Analysis
4.4. Data Quality and Metabolite Identification
4.5. Data Pre-Processing and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Risérus, U.; Willett, W.C.; Hu, F.B. Dietary fats and prevention of type 2 diabetes. Prog. Lipid Res. 2009, 48, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, R.; Seidell, J. Carbohydrate intake and obesity. Eur. J. Clin. Nutr. 2007, 61, S75–S99. [Google Scholar] [CrossRef] [PubMed]
- De Caterina, R.; Zampolli, A.; Del Turco, S.; Madonna, R.; Massaro, M. Nutritional mechanisms that influence cardiovascular disease. Am. J. Clin. Nutr. 2006, 83, 421S–426S. [Google Scholar] [CrossRef] [PubMed]
- Siri-Tarino, P.W.; Sun, Q.; Hu, F.B.; Krauss, R.M. Saturated fat, carbohydrate, and cardiovascular disease. Am. J. Clin. Nutr. 2010, 91, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Warensjö, E.; Risérus, U.; Vessby, B. Fatty acid composition of serum lipids predicts the development of the metabolic syndrome in men. Diabetologia 2005, 48, 1999–2005. [Google Scholar] [CrossRef] [PubMed]
- Vanhala, M.; Saltevo, J.; Soininen, P.; Kautiainen, H.; Kangas, A.J.; Ala-Korpela, M.; Mäntyselkä, P. Serum omega-6 polyunsaturated fatty acids and the metabolic syndrome: A longitudinal population-based cohort study. Am. J. Epidemiol. 2012, 176, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Birse, R.T.; Choi, J.; Reardon, K.; Rodriguez, J.; Graham, S.; Diop, S.; Ocorr, K.; Bodmer, R.; Oldham, S. High-fat-diet-induced obesity and heart dysfunction are regulated by the TOR pathway in Drosophila. Cell Metab. 2010, 12, 533–544. [Google Scholar] [CrossRef]
- Owusu-Ansah, E.; Perrimon, N. Modeling metabolic homeostasis and nutrient sensing in Drosophila: Implications for aging and metabolic diseases. Dis. Models Mech. 2014, 7, 343–350. [Google Scholar] [CrossRef]
- Mackay, T.F.; Anholt, R.R. Of flies and man: Drosophila as a model for human complex traits. Annu. Rev. Genomics Hum. Genet. 2006, 7, 339–367. [Google Scholar] [CrossRef]
- Beckingham, K.M.; Armstrong, J.D.; Texada, M.J.; Munjaal, R.; Baker, D.A. Drosophila melanogaster-the model organism of choice for the complex biology of multi-cellular organisms. Gravit. Space Biol. Bull. 2005, 18, 17–29. [Google Scholar]
- Jennings, B.H. Drosophila-a versatile model in biology & medicine. Mater. Today 2011, 14, 190–195. [Google Scholar]
- Wolf, M.J.; Rockman, H.A. Drosophila melanogaster as a model system for the genetics of postnatal cardiac function. Drug Discov. Today Dis. Models 2008, 5, 117–123. [Google Scholar] [CrossRef]
- Jeibmann, A.; Paulus, W. Drosophila melanogaster as a model organism of brain diseases. Int. J. Mol. Sci. 2009, 10, 407–440. [Google Scholar] [CrossRef] [PubMed]
- Prüßing, K.; Voigt, A.; Schulz, J.B. Drosophila melanogaster as a model organism for Alzheimer’s disease. Mol. Neurodegener. 2013, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.K.; Lee, K.; Zhang, Z.; Rashid, L.; Poe, A.; Hsieh, B.; Deighton, N.; Glassbrook, N.; Bodmer, R.; Gibson, G. Systems genomics of metabolic phenotypes in wild-type Drosophila melanogaster. Genetics 2014, 197, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.K.; Williams, S.; Springston, M.; Brown, J.; Freeman, K.; DesRoches, C.E.; Sokolowski, M.B.; Gibson, G. Genotype-by-diet interactions drive metabolic phenotype variation in Drosophila melanogaster. Genetics 2010, 185, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Chialvo, C.H.S.; Che, R.; Reif, D.; Motsinger-Reif, A.; Reed, L.K. Eigenvector metabolite analysis reveals dietary effects on the association among metabolite correlation patterns, gene expression, and phenotypes. Metabolomics 2016, 12, 167. [Google Scholar] [CrossRef]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef]
- Baker, M. Metabolomics: From small molecules to big ideas. Nat. Methods 2011, 8, 117–121. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Touw, W.G.; Bayjanov, J.R.; Overmars, L.; Backus, L.; Boekhorst, J.; Wels, M.; van Hijum, S.A. Data mining in the Life Sciences with Random Forest: A walk in the park or lost in the jungle? Briefings Bioinform. 2012, 14, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Cao, Y.; Zhang, Y.; Liu, J.; Bao, Y.; Wang, C.; Jia, W.; Zhao, A. Random forest in clinical metabolomics for phenotypic discrimination and biomarker selection. Evid. Based Complement. Altern. Med. 2013, 2013, 298183. [Google Scholar] [CrossRef] [PubMed]
- Krumsiek, J.; Suhre, K.; Illig, T.; Adamski, J.; Theis, F.J. Gaussian graphical modeling reconstructs pathway reactions from high-throughput metabolomics data. BMC Syst. Biol. 2011, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Dobra, A.; Hans, C.; Jones, B.; Nevins, J.R.; Yao, G.; West, M. Sparse graphical models for exploring gene expression data. J. Multivar. Anal. 2004, 90, 196–212. [Google Scholar] [CrossRef]
- Do, K.T.; Kastenmüller, G.; Mook-Kanamori, D.O.; Yousri, N.A.; Theis, F.J.; Suhre, K.; Krumsiek, J. Network-based approach for analyzing intra-and interfluid metabolite associations in human blood, urine, and saliva. J. Proteome Res. 2014, 14, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Kim, Y.; Gewirtz, A.D.; Jo, B.; Gao, C.; McDowell, I.C.; Engelhardt, B.E.; Battle, A.; Aguet, F.; Ardlie, K.G.; et al. Co-expression networks reveal the tissue-specific regulation of transcription and splicing. Genome Res. 2017, 27, 1843–1858. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2015, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The human metabolome database in 2013. Nucleic Acids Res. 2012, 41, D801–D807. [Google Scholar] [CrossRef]
- Daliri, E.B.-M.; Oh, D.H.; Lee, B.H. Bioactive peptides. Foods 2017, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Landry, B. Comparing Internet Probing Methodologies through an Analysis of Large Dynamic Graphs. Master’s Thesis, Naval Postgraduate School, Monterey, CA, USA, 2014. [Google Scholar]
- Vasudevan, D.M. Coconut Oil and Health Controversy: A Review. Int. J. Health Rehabil. Sci. (IJHRS) 2013, 2, 157–164. [Google Scholar]
- Wanders, R.J.; Komen, J.; Kemp, S. Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 2011, 278, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, N.; Kølvraa, S.; Mortensen, P.B.; Rasmussen, K. C6-C10-Dicarboxylic aciduria: Biochemical considerations in relation to diagnosis of beta-oxidation defects. Scand. J. Clin. Lab. Investig. 1982, 42, 15–27. [Google Scholar]
- Gregersen, N.; Mortensen, P.; Kølvraa, S. On the biologic origin of C 6-C 10-dicarboxylic and C 6-C 10-$ømega$-1-hydroxy monocarboxylic acids in human and rat with acyl-CoA dehydrogenation deficiencies: In vitro studies on the omega and omega-1-oxidation of medium-chain (C6-C12) fatty acids in human and rat liver. Pediatr. Res. 1983, 17, 828–834. [Google Scholar] [PubMed]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 2422–2435. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Loizides-Mangold, U.; Rando, G.; Zoete, V.; Michielin, O.; Reddy, J.K.; Wahli, W.; Riezman, H.; Thorens, B. The peroxisomal enzyme L-PBE is required to prevent the dietary toxicity of medium-chain fatty acids. Cell Rep. 2013, 5, 248–258. [Google Scholar] [CrossRef]
- Akram, M. Citric acid cycle and role of its intermediates in metabolism. Cell Biochem. Biophys. 2014, 68, 475–478. [Google Scholar] [CrossRef]
- Goto-Inoue, N.; Yamada, K.; Inagaki, A.; Furuichi, Y.; Ogino, S.; Manabe, Y.; Setou, M.; Fujii, N.L. Lipidomics analysis revealed the phospholipid compositional changes in muscle by chronic exercise and high-fat diet. Sci. Rep. 2013, 3, 3267. [Google Scholar] [CrossRef]
- Hoeks, J.; de Wilde, J.; Hulshof, M.F.; van den Berg, S.A.; Schaart, G.; van Dijk, K.W.; Smit, E.; Mariman, E.C. High fat diet-induced changes in mouse muscle mitochondrial phospholipids do not impair mitochondrial respiration despite insulin resistance. PLoS ONE 2011, 6, e27274. [Google Scholar] [CrossRef]
- Blusztajn, J.K.; Gonzalez-Coviella, I.L.; Logue, M.; Growdon, J.H.; Wurtman, R.J. Levels of phospholipid catabolic intermediates, glycerophosphocholine and glycerophosphoethanolamine, are elevated in brains of Alzheimer’s disease but not of Down’s syndrome patients. Brain Res. 1990, 536, 240–244. [Google Scholar] [CrossRef]
- Sun, L.; Hu, W.; Liu, Q.; Hao, Q.; Sun, B.; Zhang, Q.; Mao, S.; Qiao, J.; Yan, X. Metabonomics reveals plasma metabolic changes and inflammatory marker in polycystic ovary syndrome patients. J. Proteome Res. 2012, 11, 2937–2946. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, T.; Uraji, M.; Fujita, A.; Kawakami, K. Anti-oxidation activities of rice-derived peptides and their inhibitory effects on dipeptidylpeptidase-IV. Int. J. Pept. Res. Ther. 2015, 21, 479–485. [Google Scholar] [CrossRef]
- King, E.G.; Merkes, C.M.; McNeil, C.L.; Hoofer, S.R.; Sen, S.; Broman, K.W.; Long, A.D.; Macdonald, S.J. Genetic dissection of a model complex trait using the Drosophila Synthetic Population Resource. Genome Res. 2012, 22, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- King, E.G.; Macdonald, S.J.; Long, A.D. Properties and power of the Drosophila Synthetic Population Resource for the routine dissection of complex traits. Genetics 2012, 191, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.M.; DeHaven, C.D.; Barrett, T.; Mitchell, M.; Milgram, E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal. Chem. 2009, 81, 6656–6667. [Google Scholar] [CrossRef] [PubMed]
- Vulimiri, S.V.; Misra, M.; Hamm, J.T.; Mitchell, M.; Berger, A. Effects of mainstream cigarette smoke on the global metabolome of human lung epithelial cells. Chem. Res. Toxicol. 2009, 22, 492–503. [Google Scholar] [CrossRef]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef]
- DeHaven, C.D.; Evans, A.M.; Dai, H.; Lawton, K.A. Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J. Cheminform. 2010, 2, 9. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Lukasová, A. Hierarchical agglomerative clustering procedure. Pattern Recognit. 1979, 11, 365–381. [Google Scholar] [CrossRef]
- Ward, J.H., Jr. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Murtagh, F.; Legendre, P. Ward’s hierarchical clustering method: Clustering criterion and agglomerative algorithm. arXiv, 2011; arXiv:1111.6285. [Google Scholar]
- Giraud, C.; Huet, S.; Verzelen, N. Graph selection with GGMselect. Stat. Appl. Genet. Mol. Biol. 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Meinshausen, N.; Bühlmann, P. High-dimensional graphs and variable selection with the lasso. Ann. Stat. 2006, 34, 1436–1462. [Google Scholar] [CrossRef]
- Chintapalli, V.R.; Al Bratty, M.; Korzekwa, D.; Watson, D.G.; Dow, J.A. Mapping an atlas of tissue-specific Drosophila melanogaster metabolomes by high resolution mass spectrometry. PLoS ONE 2013, 8, e78066. [Google Scholar] [CrossRef] [PubMed]
- Tennessen, J.M.; Baker, K.D.; Lam, G.; Evans, J.; Thummel, C.S. The Drosophila estrogen-related receptor directs a metabolic switch that supports developmental growth. Cell Metab. 2011, 13, 139–148. [Google Scholar] [CrossRef]
- Sun, L.; Li, H.-M.; Seufferheld, M.J.; Walters, K.R., Jr.; Margam, V.M.; Jannasch, A.; Diaz, N.; Riley, C.P.; Sun, W.; Li, Y.-F.; et al. Systems-scale analysis reveals pathways involved in cellular response to methamphetamine. PLoS ONE 2011, 6, e18215. [Google Scholar] [CrossRef]
- Teets, N.M.; Peyton, J.T.; Ragland, G.J.; Colinet, H.; Renault, D.; Hahn, D.A.; Denlinger, D.L. Combined transcriptomic and metabolomic approach uncovers molecular mechanisms of cold tolerance in a temperate flesh fly. Physiol. Genom. 2012, 44, 764–777. [Google Scholar] [CrossRef]
- Heinrichsen, E.T.; Zhang, H.; Robinson, J.E.; Ngo, J.; Diop, S.; Bodmer, R.; Joiner, W.J.; Metallo, C.M.; Haddad, G.G. Metabolic and transcriptional response to a high-fat diet in Drosophila melanogaster. Mol. Metab. 2014, 3, 42–54. [Google Scholar] [CrossRef]
- Williams, S.; Dew-Budd, K.; Davis, K.; Anderson, J.; Bishop, R.; Freeman, K.; Davis, D.; Bray, K.; Perkins, L.; Hubickey, J.; et al. Metabolomic and gene expression profiles exhibit modular genetic and dietary structure linking metabolic syndrome phenotypes in Drosophila. G3: Genes Genomes Genet. 2015, 5, 2817–2829. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, H.A.; Knee, J.M.; Dennis, A.B.; Udaka, H.; Marshall, K.E.; Merritt, T.J.; Sinclair, B.J. Cold acclimation wholly reorganizes the Drosophila melanogaster transcriptome and metabolome. Sci. Rep. 2016, 6, 28999. [Google Scholar] [CrossRef] [PubMed]
- Musselman, L.P.; Fink, J.L.; Baranski, T.J. CoA protects against the deleterious effects of caloric overload in Drosophila. J. Lipid Res. 2016, 57, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2015, 44, D471–D480. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Ressom, H.W.; Wang, A.; Xuan, J.; Liu, M.C.; Gehan, E.A.; Wang, Y. The properties of high-dimensional data spaces: Implications for exploring gene and protein expression data. Nat. Rev. Cancer 2008, 8, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y. The biological significance of omega-oxidation of fatty acids. Proc. Jpn. Acad. Ser. B 2013, 89, 370–382. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oza, V.H.; Aicher, J.K.; Reed, L.K. Random Forest Analysis of Untargeted Metabolomics Data Suggests Increased Use of Omega Fatty Acid Oxidation Pathway in Drosophila Melanogaster Larvae Fed a Medium Chain Fatty Acid Rich High-Fat Diet. Metabolites 2019, 9, 5. https://doi.org/10.3390/metabo9010005
Oza VH, Aicher JK, Reed LK. Random Forest Analysis of Untargeted Metabolomics Data Suggests Increased Use of Omega Fatty Acid Oxidation Pathway in Drosophila Melanogaster Larvae Fed a Medium Chain Fatty Acid Rich High-Fat Diet. Metabolites. 2019; 9(1):5. https://doi.org/10.3390/metabo9010005
Chicago/Turabian StyleOza, Vishal H., Joseph K. Aicher, and Laura K. Reed. 2019. "Random Forest Analysis of Untargeted Metabolomics Data Suggests Increased Use of Omega Fatty Acid Oxidation Pathway in Drosophila Melanogaster Larvae Fed a Medium Chain Fatty Acid Rich High-Fat Diet" Metabolites 9, no. 1: 5. https://doi.org/10.3390/metabo9010005
APA StyleOza, V. H., Aicher, J. K., & Reed, L. K. (2019). Random Forest Analysis of Untargeted Metabolomics Data Suggests Increased Use of Omega Fatty Acid Oxidation Pathway in Drosophila Melanogaster Larvae Fed a Medium Chain Fatty Acid Rich High-Fat Diet. Metabolites, 9(1), 5. https://doi.org/10.3390/metabo9010005