Urolithin A Is a Dietary Microbiota-Derived Human Aryl Hydrocarbon Receptor Antagonist




Abstract
1. Introduction
2. Results
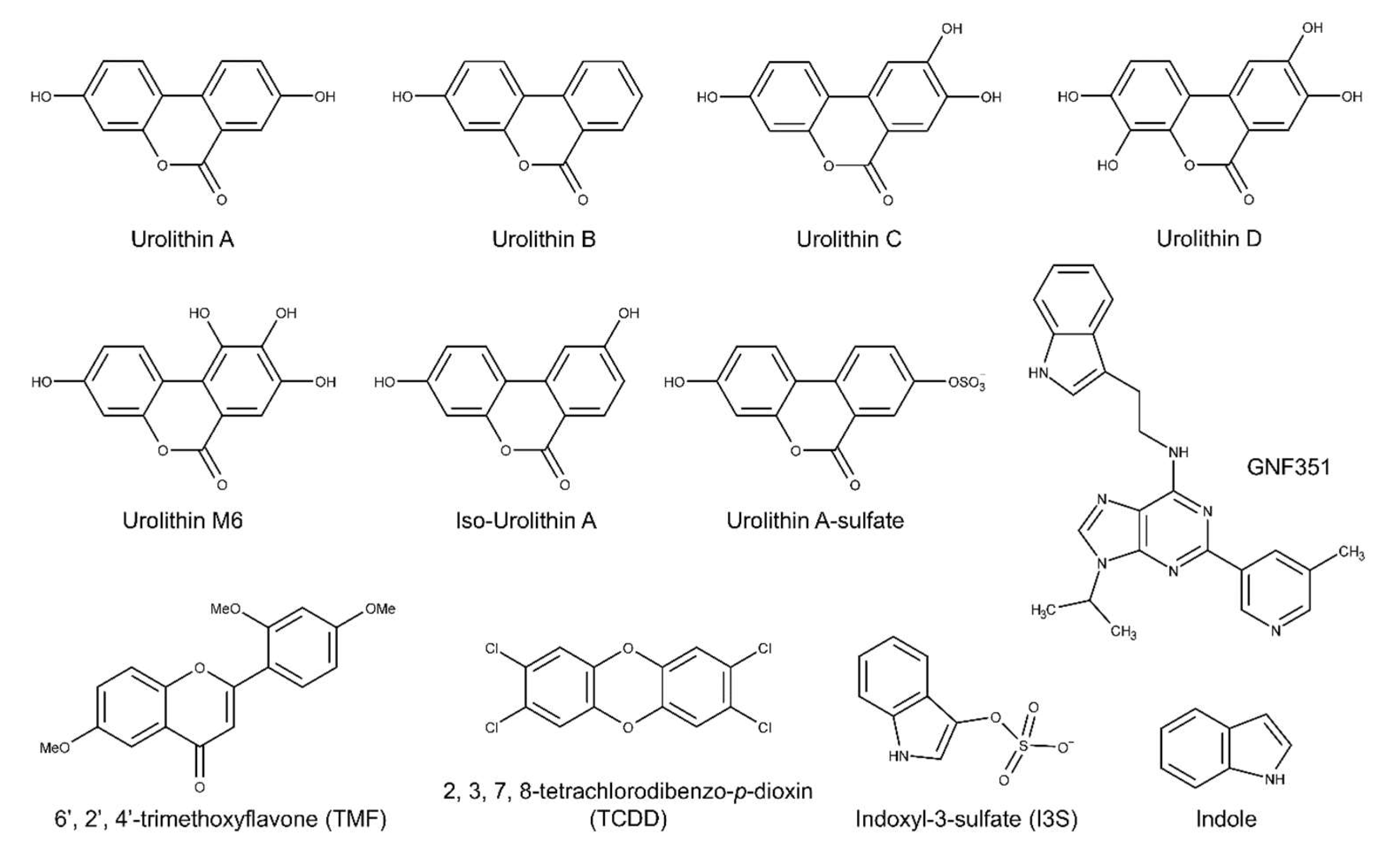
2.1. Urolithin Metabolites Do Not Activate AHR-Dependent DRE-Mediated Transcription
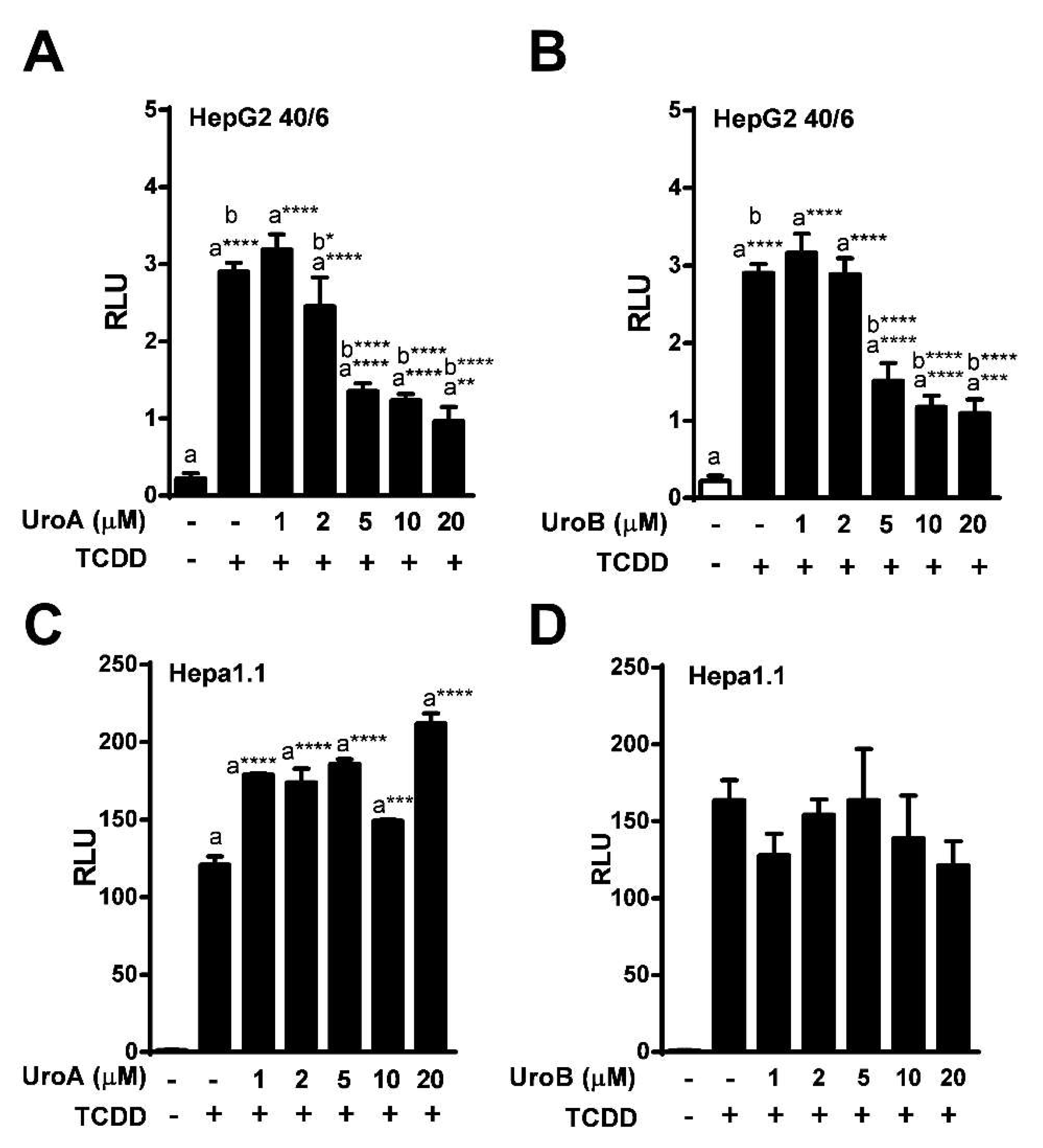
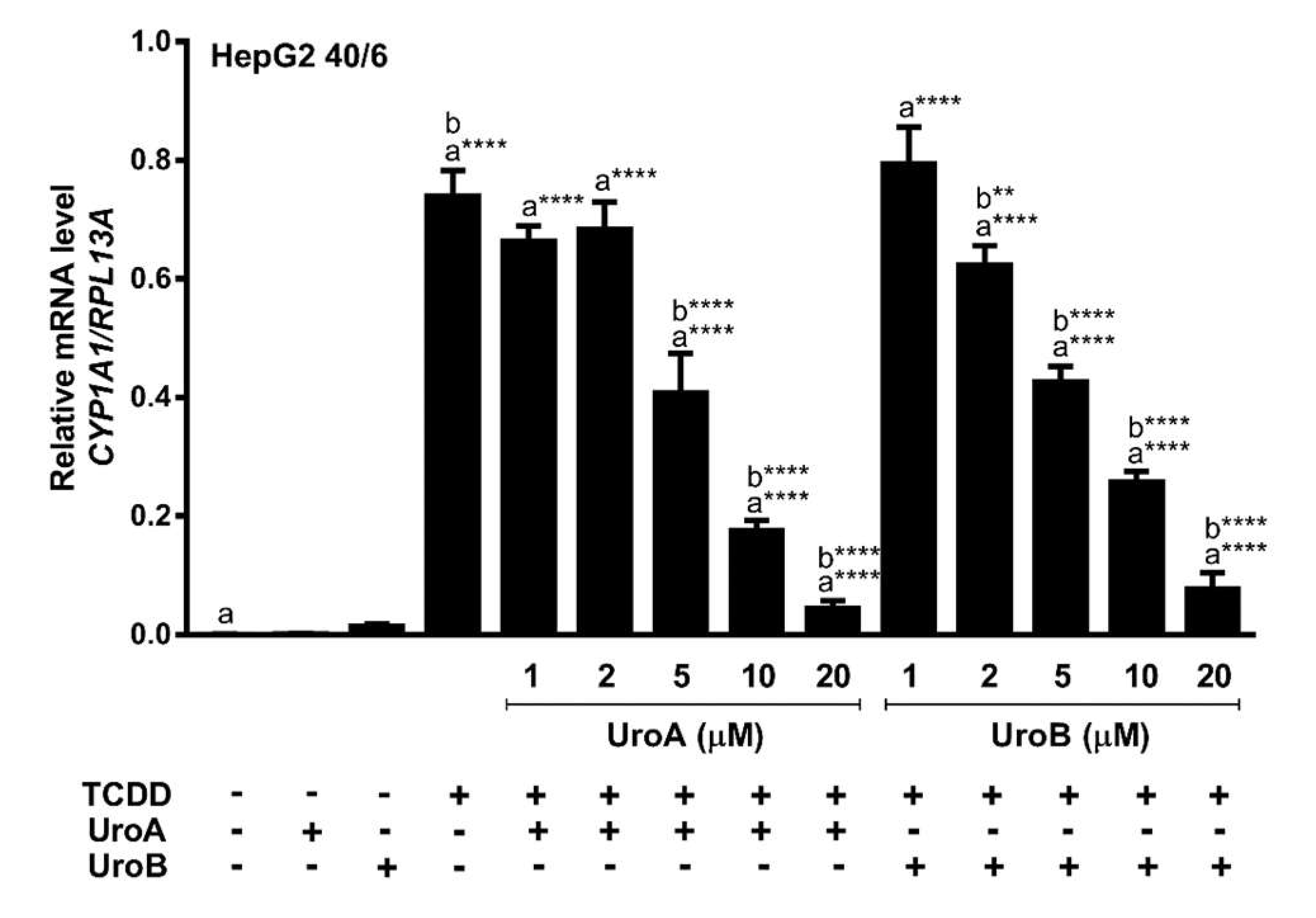
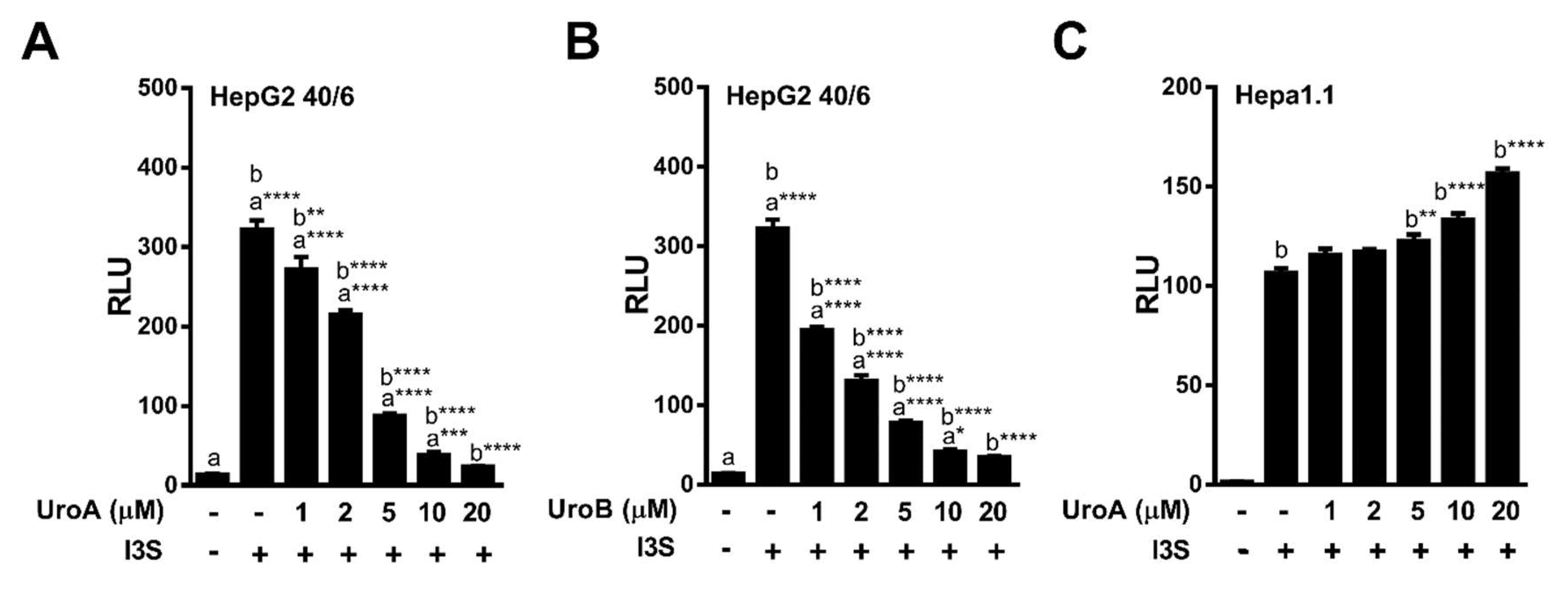
2.2. Urolithins A, B, C Antagonize Ligand-Mediated AHR Transcriptional Activity in a Human-Selective Manner
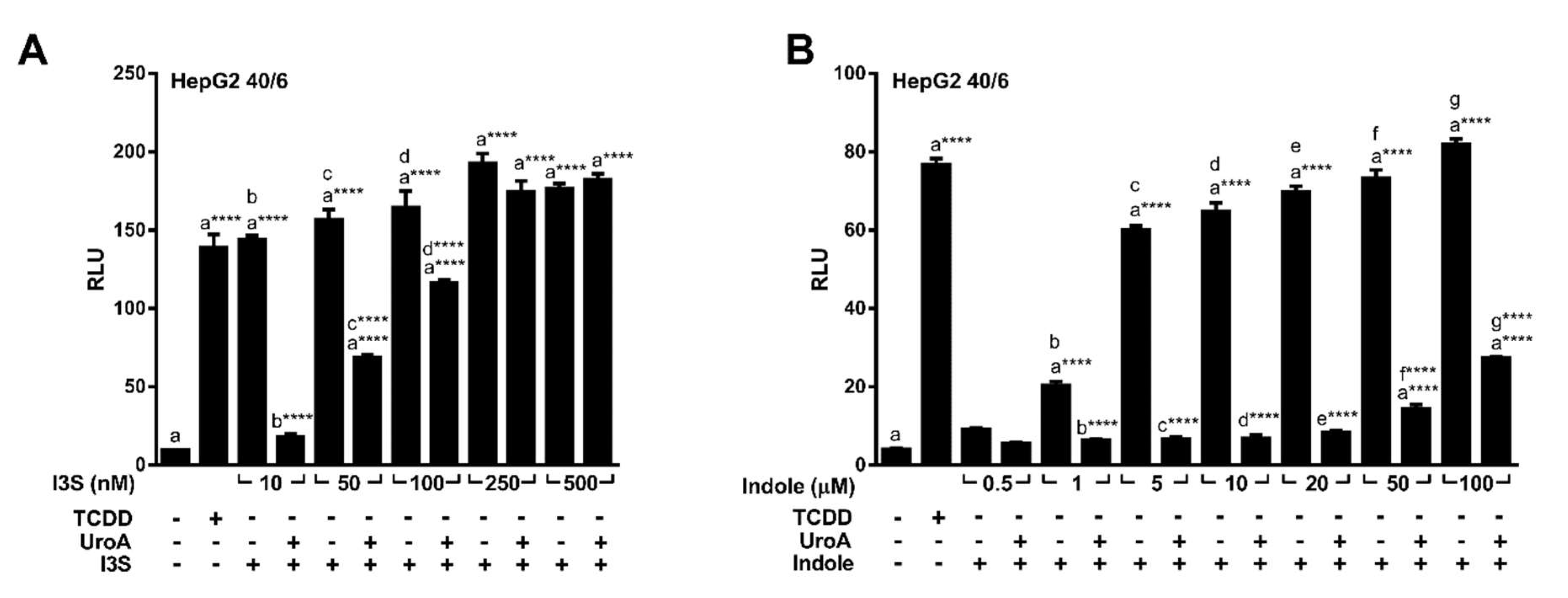
2.3. Endogenous Ligands I3S and Indole Are Antagonized by Urolithin A
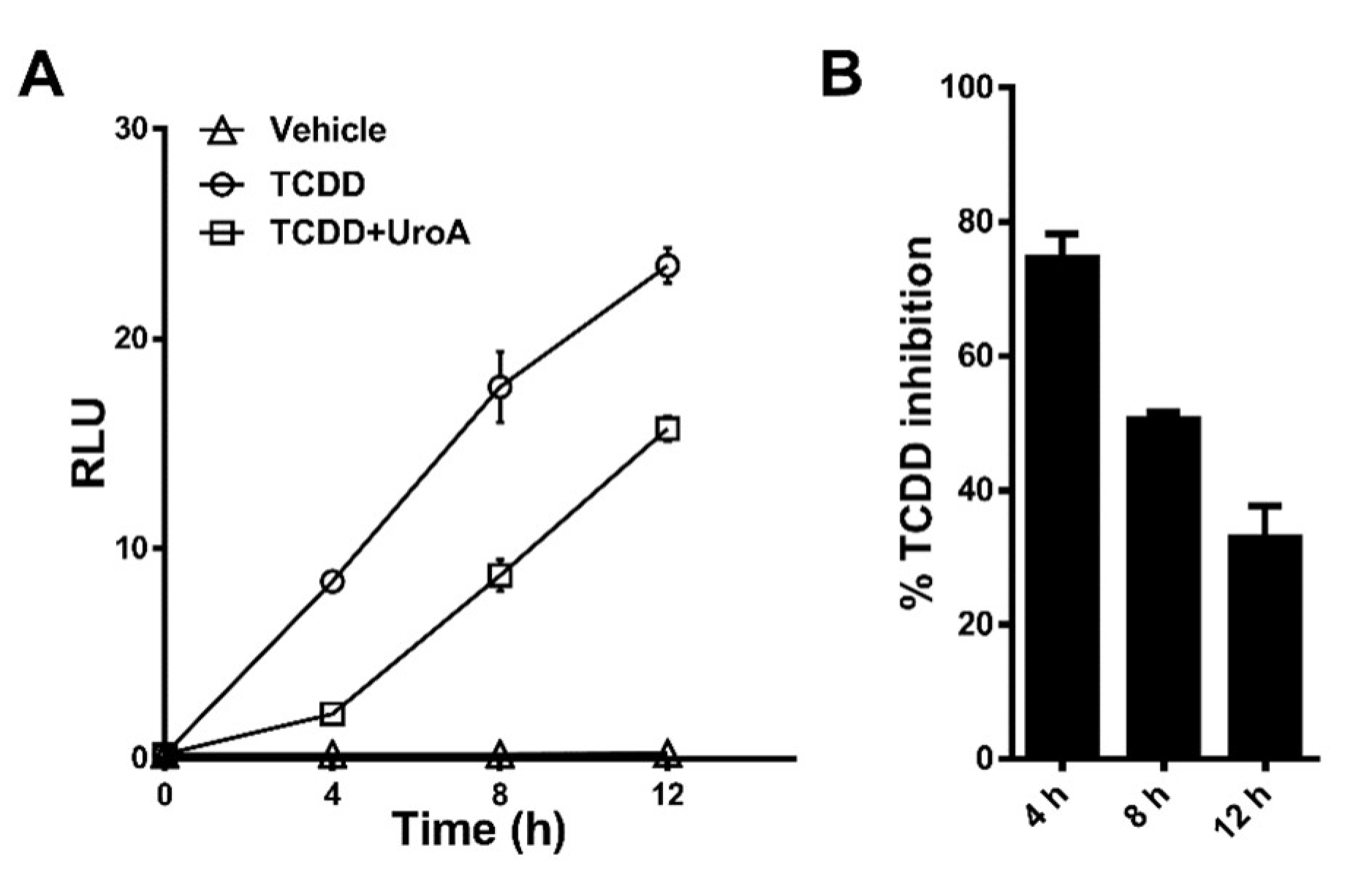
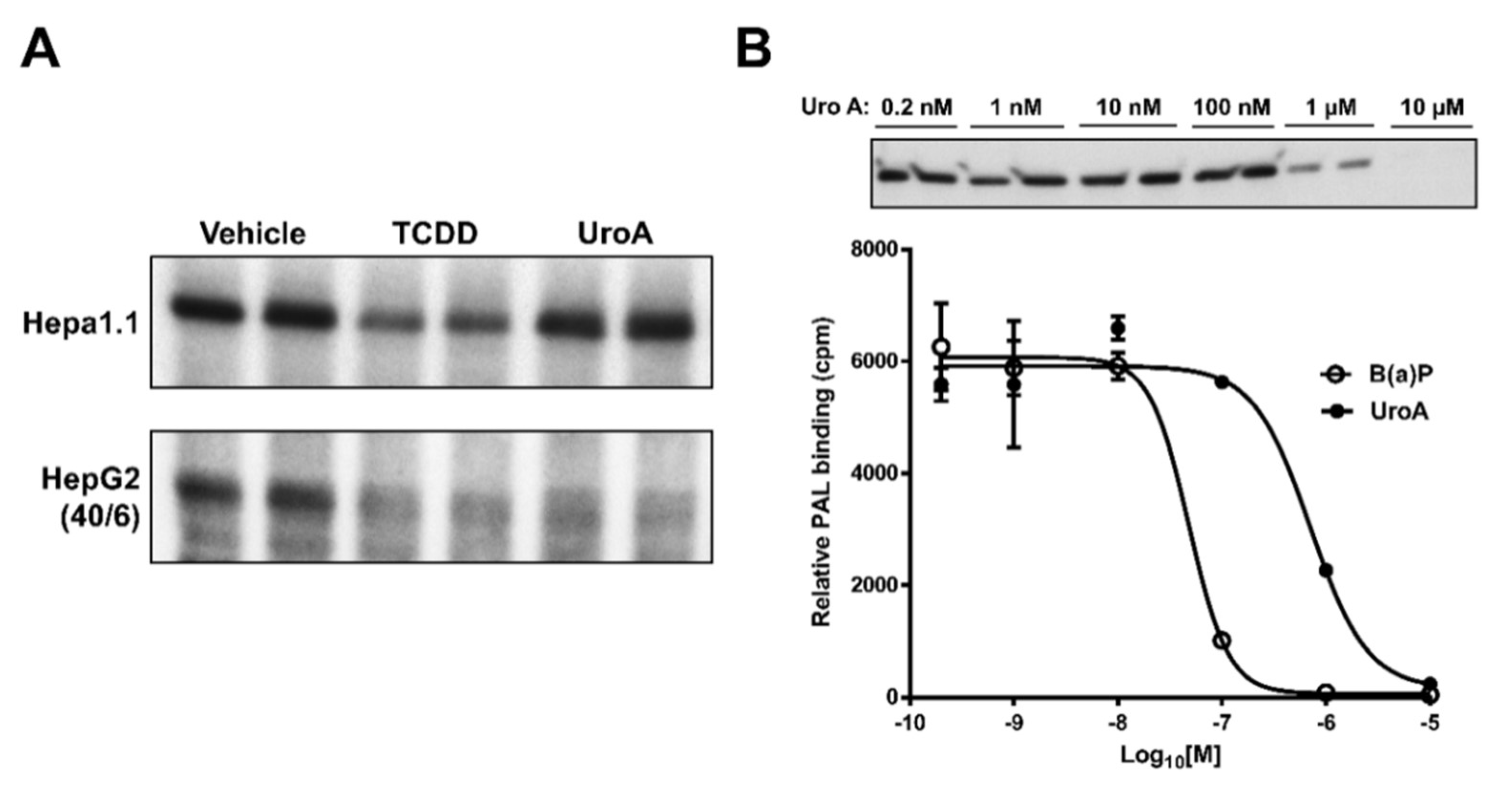
2.4. Urolithin A Exhibits Sustained Antagonism
2.5. Urolithin A is a Direct AHR Ligand
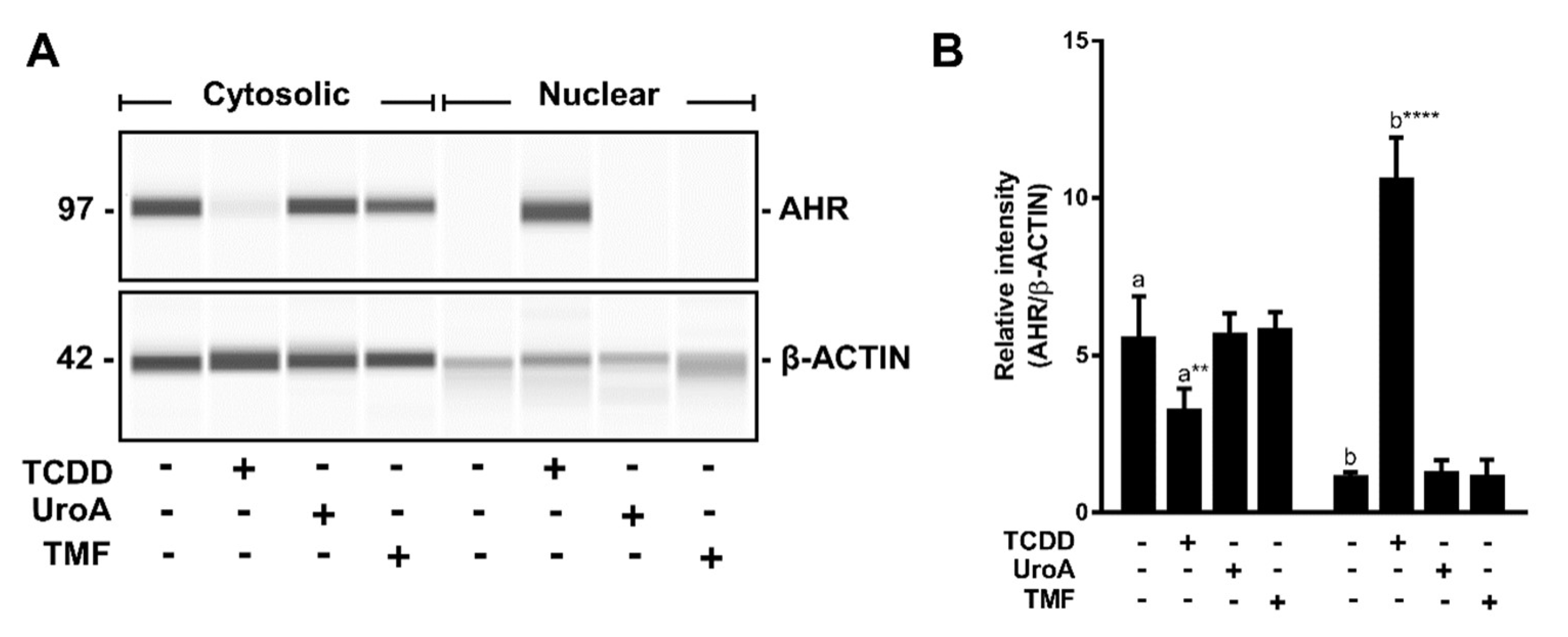
2.6. Urolithin A Does Not Induce AHR Retention in the Nucleus
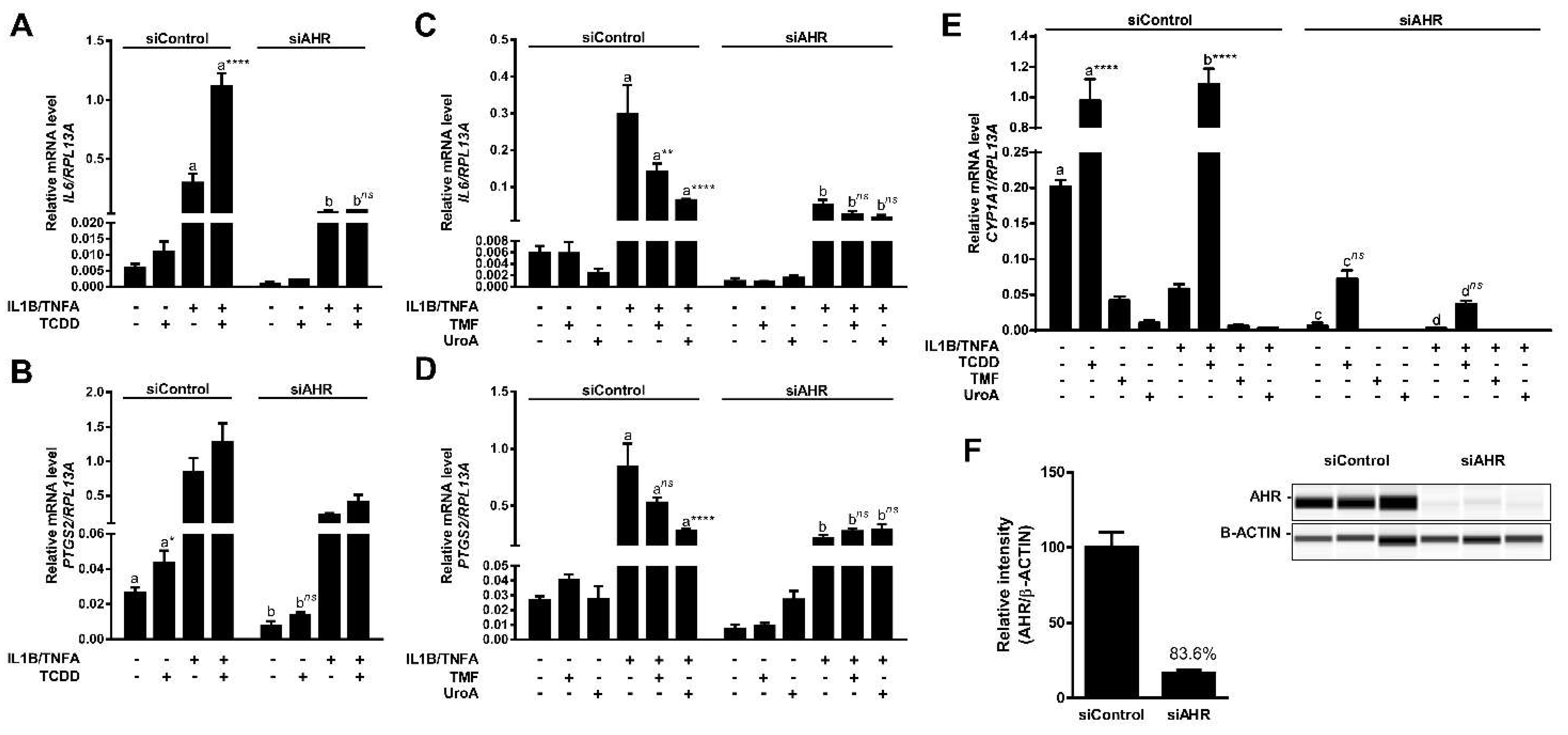
2.7. Urolithin A Attenuates Cytokine-Induced Inflammatory Signaling in an AHR-Dependent Manner
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Luciferase Reporter Assays
4.4. PAL Ligand Competition Assay
4.5. RNA Isolation and Quantitative Real-Time PCR Analysis
4.6. AHR Nuclear Translocation Analysis
4.7. Gene Silencing
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Durrin, L.K.; Jones, P.B.C.; Fisher, J.M.; Galeazzi, D.R.; Whitlock, J.P., Jr. 2,3,7,8-Tetrachlorodibenzo-p-dioxin receptors regulate transcription of the cytochrome P1-450 gene. J. Cell. Biochem. 1987, 35, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, J.C.; Gustafsson, J.Å. Aryl hydrocarbon receptor-mediated signal transduction. Crit. Rev. Toxicol. 1997, 27, 109–134. [Google Scholar] [CrossRef] [PubMed]
- Hao, N.; Whitelaw, M.L. The emerging roles of AhR in physiology and immunity. Biochem. Pharmacol. 2013, 86, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Perdew, G.H. Ligand activation of the Ah receptor contributes to gastrointestinal homeostasis. Curr. Opin. Toxicol. 2016, 1, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and tryptophan metabolism: Endogenous and dietary routes to Ah receptor activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Espín, J.C.; Larrosa, M.; García-Conesa, M.; Tomás-Barberán, F. Biological significance of the gut microbial ellagic acid-derived metabolites urolithins. Evid.-Based Complement. Altern. Med. 2013, 2013, 1–15. [Google Scholar] [CrossRef] [PubMed]
- González-Sarrías, A.; García-Villalba, R.; Núñez-Sánchez, M.Á.; Tomé-Carneiro, J.; Zafrilla, P.; Mulero, J.; Tomás-Barberán, F.A.; Espín, J.C. Identifying the limits for ellagic acid bioavailability: A crossover pharmacokinetic study in healthy volunteers after consumption of pomegranate extracts. J. Funct. Foods 2015, 19, 225–235. [Google Scholar] [CrossRef]
- Cerdá, B.; Tomás-Barberán, F.A.; Espín, J.C. Metabolism of antioxidant and chemopreventive ellagitannins from strawberries, raspberries, walnuts, and oak-aged wine in humans: Identification of biomarkers and individual variability. J. Agric. Food Chem. 2005, 53, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Nuñez-Sánchez, M.A.; García-Villalba, R.; Monedero-Saiz, T.; García-Talavera, N.V.; Gómez-Sánchez, M.B.; Sánchez-Álvarez, C.; García-Albert, A.M.; Rodríguez-Gil, F.J.; Ruiz-Marín, M.; Pastor-Quirante, F.A.; et al. Targeted metabolic profiling of pomegranate polyphenols and urolithins in plasma, urine and colon tissues from colorectal cancer patients. Mol. Nutr. Food Res. 2014, 58, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Cerdá, B.; Espín, J.C.; Parra, S.; Martínez, P.; Tomás-Barberán, F.A. The potent in vitro antioxidant ellagitannins from pomegranate juice are metabolised into bioavailable but poor antioxidant hydroxy-6H-dibenzopyran-6-one derivatives by the colonic microflora of healthy humans. Eur. J. Nutr. 2004, 43, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Seeram, N.P.; Henning, S.M.; Zhang, Y.; Suchard, M.; Li, Z.; Heber, D. Pomegranate juice ellagitannin metabolites are present in human plasma and some persist in urine for up to 48 hours. J. Nutr. 2006, 136, 2481–2485. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Barberan, F.A.; Espín, J.C.; García-Conesa, M.T. Bioavailability and metabolism of ellagic acid and ellagitannins. In Chemistry and Biology of Ellagitannins; World Scientific: Singapore, 2009; pp. 273–297. [Google Scholar]
- Cerdá, B.; Periago, P.; Espín, J.C.; Tomás-Barberán, F.A. Identification of Urolithin A as a metabolite produced by human colon microflora from ellagic acid and related compounds. J. Agric. Food Chem. 2005, 53, 5571–5576. [Google Scholar] [CrossRef] [PubMed]
- Larrosa, M.; García-Conesa, M.T.; Espín, J.C.; Tomás-Barberán, F.A. Ellagitannins, ellagic acid and vascular health. Mol. Asp. Med. 2010, 31, 513–539. [Google Scholar] [CrossRef] [PubMed]
- Heber, D. Multitargeted therapy of cancer by ellagitannins. Cancer Lett. 2008, 269, 262–268. [Google Scholar] [CrossRef] [PubMed]
- González-Sarrías, A.; Azorín-Ortuño, M.; Yáñez-Gascón, M.J.; Tomás-Barberán, F.A.; García-Conesa, M.T.; Espín, J.C. Dissimilar in vitro and in vivo effects of ellagic acid and its microbiota-derived metabolites, urolithins, on the cytochrome P450 1A1. J. Agric. Food Chem. 2009, 57, 5623–5632. [Google Scholar] [CrossRef]
- Ikuta, T.; Kurosumi, M.; Yatsuoka, T.; Nishimura, Y. Tissue distribution of aryl hydrocarbon receptor in the intestine: Implication of putative roles in tumor suppression. Exp. Cell Res. 2016, 343, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Muku, G.E.; Lahoti, T.S.; Murray, I.A.; Podolsky, M.A.; Smith, K.J.; Hubbard, T.D.; Kuzu, G.; Gowda, K.; Amin, S.G.; Perdew, G.H. Ligand-mediated cytoplasmic retention of the Ah receptor inhibits macrophage-mediated acute inflammatory responses. Lab. Investig. 2017, 97, 1471–1487. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Krishnegowda, G.; DiNatale, B.C.; Flaveny, C.; Chiaro, C.; Lin, J.M.; Sharma, A.K.; Amin, S.; Perdew, G.H. Development of a selective modulator of aryl hydrocarbon (Ah) receptor activity that exhibits anti-inflammatory properties. Chem. Res. Toxicol. 2010, 23, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C. The AHR in the control of cell cycle and apoptosis. In The Ah Receptor in Biology and Toxicology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 467–483. [Google Scholar]
- Tomás-Barberán, F.A.; González-Sarrías, A.; García-Villalba, R.; Núñez-Sánchez, M.A.; Selma, M.V.; García-Conesa, M.T.; Espín, J.C. Urolithins, the rescue of “old” metabolites to understand a “new” concept: Metabotypes as a nexus among phenolic metabolism, microbiota dysbiosis, and host health status. Mol. Nutr. Food Res. 2017, 61, 1500901. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Schroeder, J.C.; Perdew, G.H. Ah receptor antagonism inhibits constitutive and cytokine inducible IL6 production in head and neck tumor cell lines. Mol. Carcinog. 2011, 50, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Lahoti, T.S.; John, K.; Hughes, J.M.; Kusnadi, A.; Murray, I.A.; Krishnegowda, G.; Amin, S.; Perdew, G.H. Aryl hydrocarbon receptor antagonism mitigates cytokine-mediated inflammatory signalling in primary human fibroblast-like synoviocytes. Ann. Rheum. Dis. 2013, 72, 1708–1716. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, T.D.; Murray, I.A.; Bisson, W.H.; Lahoti, T.S.; Gowda, K.; Amin, S.G.; Patterson, A.D.; Perdew, G.H. Adaptation of the human aryl hydrocarbon receptor to sense microbiota-derived indoles. Sci. Rep. 2015, 5, 12689. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.C.; DiNatale, B.C.; Murray, I.A.; Flaveny, C.A.; Liu, Q.; Laurenzana, E.M.; Lin, J.M.; Strom, S.C.; Omiecinski, C.J.; Amin, S.; et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry 2010, 49, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Adachi, J.; Mori, Y.; Matsui, S.; Takigami, H.; Fujino, J.; Kitagawa, H.; Miller, C.A., III; Kato, T.; Saeki, K.; Matsuda, T. Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. J. Biol. Chem. 2001, 276, 31475–31478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qin, C.; Safe, S.H. Flavonoids as aryl hydrocarbon receptor agonists/antagonists: Effects of structure and cell context. Environ. Health Perspect. 2003, 111, 1877–1882. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Flaveny, C.A.; DiNatale, B.C.; Chairo, C.R.; Schroeder, J.C.; Kusnadi, A.; Perdew, G.H. Antagonism of aryl hydrocarbon receptor signaling by 6,2’,4’-trimethoxyflavone. J. Pharmacol. Exp. Ther. 2010, 332, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Day, A.J.; Cañada, F.J.; Díaz, J.C.; Kroon, P.A.; Mclauchlan, R.; Faulds, C.B.; Plumb, G.W.; Morgan, M.R.; Williamson, G. Dietary flavonoid and isoflavone glycosides are hydrolysed by the lactase site of lactase phlorizin hydrolase. FEBS Lett. 2010, 468, 166–170. [Google Scholar] [CrossRef]
- Day, A.J.; DuPont, M.S.; Ridley, S.; Rhodes, M.; Rhodes, M.J.; Morgan, M.R.; Williamson, G. Deglycosylation of flavonoid and isoflavonoid glycosides by human small intestine and liver β-glucosidase activity. FEBS Lett. 1998, 436, 71–75. [Google Scholar] [CrossRef]
- Bang, S.H. Metabolism of rutin and poncirin by human intestinal microbiota and cloning of their metabolizing α-L-rhamnosidase from Bifidobacterium dentium. J. Microbiol. Biotechnol. 2015, 25, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Doyle, B.; Griffiths, L.A. The metabolism of ellagic acid in the rat. Xenobiotica 1980, 10, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Espín, J.C.; González-Barrio, R.; Cerdá, B.; López-Bote, C.; Rey, A.I.; Tomás-Barberán, F.A. Iberian pig as a model to clarify obscure points in the bioavailability and metabolism of ellagitannins in humans. J. Agric. Food Chem. 2007, 55, 10476–10485. [Google Scholar] [CrossRef] [PubMed]
- González-Sarrías, A.; García-Villalba, R.; Romo-Vaquero, M.; Alasalvar, C.; Örem, A.; Zafrilla, P.; Tomás-Barberán, F.A.; Selma, M.V.; Espín, J.C. Clustering according to urolithin metabotype explains the interindividual variability in the improvement of cardiovascular risk biomarkers in overweight-obese individuals consuming pomegranate: A randomized clinical trial. Mol. Nutr. Food Res. 2017, 61, 1600830. [Google Scholar] [CrossRef] [PubMed]
- Larrosa, M.; González-Sarrías, A.; Yáñez-Gascón, M.J.; Selma, M.V.; Azorín-Ortuño, M.; Toti, S.; Tomás-Barberán, F.; Dolara, P.; Espín, J.C. Anti-inflammatory properties of a pomegranate extract and its metabolite urolithin-A in a colitis rat model and the effect of colon inflammation on phenolic metabolism. J. Nutr. Biochem. 2010, 21, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, H.; Shibata, M.; Myojin, Y.; Ito, H.; Sugimoto, Y.; Tai, A.; Hatano, T. In vivo anti-inflammatory and antioxidant properties of ellagitannin metabolite urolithin A. Bioorg. Med. Chem. Lett. 2011, 21, 5901–5904. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.J.; Boyer, J.A.; Muku, G.E.; Murray, I.A.; Gowda, K.; Desai, D.; Amin, S.G.; Glick, A.B.; Perdew, G.H. Editor’s Highlight: Ah receptor activation potentiates neutrophil chemoattractant (C-X-C Motif) ligand 5 expression in keratinocytes and skin. Toxicol. Sci. 2017, 160, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Lahoti, T.S.; Boyer, J.A.; Kusnadi, A.; Muku, G.E.; Murray, I.A.; Perdew, G.H. Aryl hydrocarbon receptor activation synergistically induces lipopolysaccharide-mediated expression of proinflammatory chemokine (c–c motif) ligand 20. Toxicol. Sci. 2015, 148, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Bleich, D. Transcriptional regulation of cyclooxygenase-2 gene in pancreatic β-cells. J. Biol. Chem. 2004, 279, 35403–35411. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Kang, H.; Song, C.; Lei, Z.; Li, L.; Guo, J.; Xu, Y.; Guan, H.; Fang, Z.; Li, F. Urolithin A Inhibits the Catabolic Effect of TNFα on Nucleus Pulposus Cell and Alleviates Intervertebral Disc Degeneration in vivo. Front. Pharmacol. 2018, 9, 1043. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, W.; Kishi, H.; Yagasaki, K.; Ohhira, S. Urolithin A attenuates pro-inflammatory mediator production by suppressing PI3-K/Akt/NF-κB and JNK/AP-1 signaling pathways in lipopolysaccharide-stimulated RAW264 macrophages: Possible involvement of NADPH oxidase-derived reactive oxygen species. Eur. J. Pharmacol. 2018, 833, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yuan, C.; Wang, G.; Luo, J.; Ma, H.; Xu, L.; Mu, Y.; Li, Y.; Seeram, N.P.; Huang, X.; et al. Urolithins Attenuate LPS-Induced Neuroinflammation in BV2 Microglia via MAPK, Akt, and NF-κB Signaling Pathways. J. Agric. Food Chem. 2018, 66, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Kojadinovic, M.; Arsic, A.; Petovic-Oggiano, G.; Gavrovic-Jankulovic, M.; Glibetic, M.; Popovic, M. Effect of urolithins on oxidative stress of colorectal adenocarcinomacells-Caco-2. Int. J. Food Sci. Nutr. 2017, 68, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Lahvis, G.P.; Lindell, S.L.; Thomas, R.S.; McCuskey, R.S.; Murphy, C.; Glover, E.; Bentz, M.; Southard, J.; Bradfield, C.A. Portosystemic shunting and persistent fetal vascular structures in aryl hydrocarbon receptor-deficient mice. Proc. Natl. Acad. Sci. USA 2000, 97, 10442–10447. [Google Scholar] [CrossRef] [PubMed]
- Lahvis, G.P.; Pyzalski, R.W.; Glover, E.; Pitot, H.C.; McElwee, M.K.; Bradfield, C.A. The aryl hydrocarbon receptor is required for developmental closure of the ductus venosus in the neonatal mouse. Mol. Pharmacol. 2004, 67, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Salguero, P.; Pineau, T.; Hilbert, D.M.; McPhail, T.; Lee, S.S.; Kimura, S.; Nebert, D.W.; Rudikoff, S.; Ward, J.M.; Gonzalez, F.J. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 1995, 268, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Mimura, J.; Nakamura, N.; Harada, N.; Yamamoto, M.; Morohashi, K.; Fujii-Kuriyama, Y. Intrinsic function of the aryl hydrocarbon (dioxin) receptor as a key factor in female reproduction. Mol. Cell. Biol. 2005, 25, 10040–10051. [Google Scholar] [CrossRef] [PubMed]
- Andersson, P.; McGuire, J.; Rubio, C.; Gradin, K.; Whitelaw, M.L.; Pettersson, S.; Hanberg, A.; Poellinger, L. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 9990–9995. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Novikov, O.; Wang, Z.; Stanford, E.A.; Parks, A.J.; Ramirez-Cardenas, A.; Landesman, E.; Laklouk, I.; Sarita-Reyes, C.; Gusenleitner, D.; Li, A.; et al. An aryl hydrocarbon receptor-mediated amplification loop that enforces cell migration in ER-/PR-/Her2-human breast cancer cells. Mol. Pharmacol. 2016, 90, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Stanford, E.A.; Ramirez-Cardenas, A.; Wang, Z.; Novikov, O.; Alamoud, K.; Koutrakis, P.; Mizgerd, J.P.; Genco, C.A.; Kukuruzinska, M.; Monti, S.; et al. Role for the aryl hydrocarbon receptor and diverse ligands in oral squamous cell carcinoma migration and tumorigenesis. Mol. Cancer Res. 2016, 14, 696–706. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Smith, K.; John, K.; Krishnegowda, G.; Amin, S.G.; Perdew, G.H. Ah receptor antagonism represses head and neck tumor cell aggressive phenotype. Mol. Cancer Res. 2012, 10, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- John, K.; Lahoti, T.S.; Wagner, K.; Hughes, J.M.; Perdew, G.H. The Ah receptor regulates growth factor expression in head and neck squamous cell carcinoma cell lines. Mol. Carcinog. 2014, 53, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Flaveny, C.A.; Murray, I.A.; Chiaro, C.R.; Perdew, G.H. Ligand selectivity and gene regulation by the human aryl hydrocarbon receptor in transgenic mice. Mol. Pharmacol. 2009, 75, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, T.D.; Murray, I.A.; Bisson, W.H.; Sullivan, A.P.; Sebastian, A.; Perry, G.H.; Jablonski, N.G.; Perdew, G.H. Divergent Ah receptor ligand selectivity during hominin evolution. Mol. Biol. Evol. 2016, 33, 2648–2658. [Google Scholar] [CrossRef] [PubMed]
- Faber, S.C.; Soshilov, A.A.; Giani Tagliabue, S.; Bonati, L.; Denison, M.S. Comparative In Vitro and In Silico Analysis of the Selectivity of Indirubin as a Human Ah Receptor Agonist. Int. J. Mol. Sci. 2018, 19, 2692. [Google Scholar] [CrossRef] [PubMed]
- González-Sarrías, A.; Espín, J.-C.; Tomás-Barberán, F.A.; García-Conesa, M.-T. Gene expression, cell cycle arrest and MAPK signalling regulation in Caco-2 cells exposed to ellagic acid and its metabolites, urolithins. Mol. Nutr. Food Res. 2009, 53, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Li, L.; Celver, J.; Killian, C.; Kovoor, A.; Seeram, N.P. Effects of fruit ellagitannin extracts, ellagic acid, and their colonic metabolite, Urolithin A, on Wnt signaling. J. Agric. Food Chem. 2010, 58, 3965–3969. [Google Scholar] [CrossRef] [PubMed]
- González-Sarrías, A.; Núñez-Sánchez, M.Á.; Tomé-Carneiro, J.; Tomás-Barberán, F.A.; García-Conesa, M.T.; Espín, J.C. Comprehensive characterization of the effects of ellagic acid and urolithins on colorectal cancer and key-associated molecular hallmarks: MicroRNA cell specific induction of CDKN1A (p21) as a common mechanism involved. Mol. Nutr. Food Res. 2016, 60, 701–716. [Google Scholar] [CrossRef] [PubMed]
- Kasimsetty, S.G.; Bialonska, D.; Reddy, M.K.; Ma, G.; Khan, S.I.; Ferreira, D. Colon cancer chemopreventive activities of pomegranate ellagitannins and urolithins. J. Agric. Food Chem. 2010, 58, 2180–2187. [Google Scholar] [CrossRef] [PubMed]
- Nuñez-Sánchez, M.A.; Dávalos, A.; González-Sarrías, A.; Casas-Agustench, P.; Visioli, F.; Monedero-Saiz, T.; García-Talavera, N.V.; Gómez-Sánchez, M.B.; Sánchez-Álvarez, C.; García-Albert, A.M.; et al. MicroRNAs expression in normal and malignant colon tissues as biomarkers of colorectal cancer and in response to pomegranate extracts consumption: Critical issues to discern between modulatory effects and potential artefacts. Mol. Nutr. Food Res. 2015, 59, 1973–1986. [Google Scholar] [CrossRef] [PubMed]
- Totiger, T.M.; Srinivasan, S.; Jala, V.R.; Lamichhane, P.; Dosch, A.R.; Gaidarski, A.A.; Joshi, C.; Rangappa, S.; Castellanos, J.; Vemula, P.K.; et al. Urolithin A, a novel natural compound to target PI3K/AKT/mTOR pathway in pancreatic cancer. Mol. Cancer Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, N.R.; Chandrasekaran, B.; Kolluru, V.; Ankem, M.; Damodaran, C.; Vadhanam, M.V. A natural molecule, urolithin A, downregulates androgen receptor activation and suppresses growth of prostate cancer. Mol. Carcinog. 2018, 57, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- García-Villalba, R.; Espín, J.C.; Tomás-Barberán, F.A. Chromatographic and spectroscopic characterization of urolithins for their determination in biological samples after the intake of foods containing ellagitannins and ellagic acid. J. Chromatogr. A 2016, 1428, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Long, W.P.; Pray-Grant, M.; Tsai, J.C.; Perdew, G.H. Protein kinase C activity is required for aryl hydrocarbon receptor pathway-mediated signal transduction. Mol. Pharmacol. 1998, 53, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Hord, N.G.; Perdew, G.H. Characterization of the activated form of the aryl hydrocarbon receptor in the nucleus of HeLa cells in the absence of exogenous ligand. Arch. Biochem. Biophys. 1996, 329, 47–55. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence |
|---|---|
| CYP1A1 | 5′-TACCTCAGCCACCTCCAAGAT-3′ 5′-GAGGTCTTGAGGCCCTGAT-3′ |
| IL6 | 5′-AAATTCGGTACATCCTCGACG-3′ 5′-AGTGCCTCTTTGCTGCTTTCA-3′ |
| PTGS2 | 5′-CAAATCCTTGCTGTTCCCACCCAT-3′ 5′-GTGCACTGTGTTTGGAGTGGGTTT-3′ |
| RPL13A | 5′-CCTGGAGGAGAAGAGGAAAGAGA-3′ 5′-GAGGACCTCTGTGTATTTGTCA-3′ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muku, G.E.; Murray, I.A.; Espín, J.C.; Perdew, G.H. Urolithin A Is a Dietary Microbiota-Derived Human Aryl Hydrocarbon Receptor Antagonist. Metabolites 2018, 8, 86. https://doi.org/10.3390/metabo8040086
Muku GE, Murray IA, Espín JC, Perdew GH. Urolithin A Is a Dietary Microbiota-Derived Human Aryl Hydrocarbon Receptor Antagonist. Metabolites. 2018; 8(4):86. https://doi.org/10.3390/metabo8040086
Chicago/Turabian StyleMuku, Gulsum E., Iain A. Murray, Juan C. Espín, and Gary H. Perdew. 2018. "Urolithin A Is a Dietary Microbiota-Derived Human Aryl Hydrocarbon Receptor Antagonist" Metabolites 8, no. 4: 86. https://doi.org/10.3390/metabo8040086
APA StyleMuku, G. E., Murray, I. A., Espín, J. C., & Perdew, G. H. (2018). Urolithin A Is a Dietary Microbiota-Derived Human Aryl Hydrocarbon Receptor Antagonist. Metabolites, 8(4), 86. https://doi.org/10.3390/metabo8040086