A Framework for Development of Useful Metabolomic Biomarkers and Their Effective Knowledge Translation


Abstract
1. Introduction
1.1. Identifying the Clinical Application: the Prime Objective
1.2. The Biomarker Development Process: Setting the Stage for Effective Knowledge Translation
2. Sample Considerations
3. Selection of Analytical Platform
4. Assay Design
5. Patent Protection and Regulatory Approval
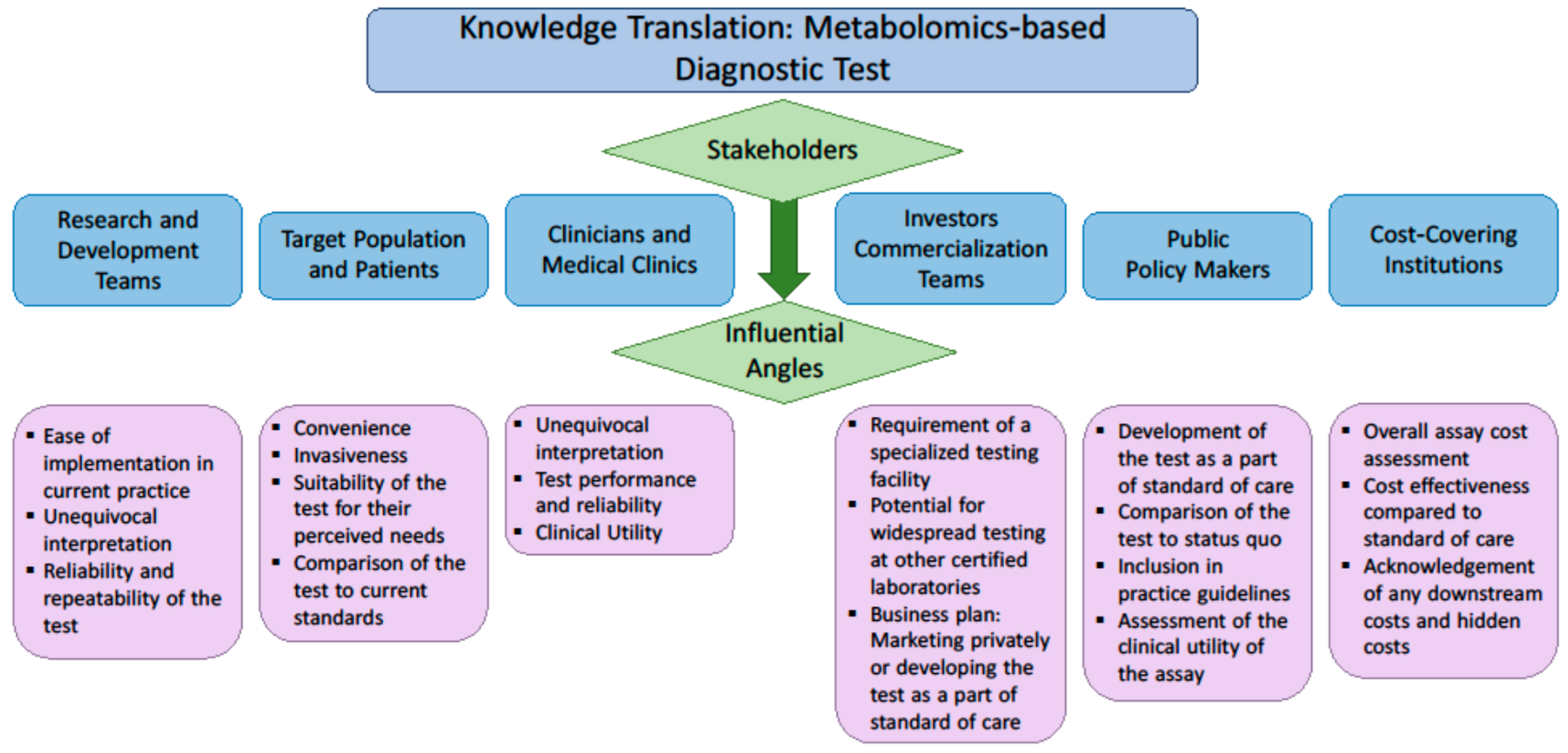
6. Knowledge Translation: Engagement of the Stakeholders
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 1532–6535. [Google Scholar]
- Ginsburg, G.S.; Willard, H.F. Genomic and personalized medicine: Foundations and applications. Transl. Res. 2009, 154, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.M.; Ferguson, J.P.; Zheng, W.; Qian, F.; Bruno, V.; Montgomery, R.R.; Zhao, H. Differential expression analysis for paired rna-seq data. BMC Bioinform. 2013, 14, 110. [Google Scholar] [CrossRef] [PubMed]
- McGettigan, P.A. Transcriptomics in the rna-seq era. Curr. Opin. Chem. Biol. 2013, 17, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.W.; Kim, H.J.; Lee, Y.S.; Myong, N.H.; Hwang, C.H.; Lee, G.S.; Yom, H.C. The proteomics approach to find biomarkers in gastric cancer. J. Korean Med. Sci. 2003, 18, 505. [Google Scholar] [CrossRef] [PubMed]
- Moscow, J.A.; Cowan, K.H. Multidrug resistance1. JNCI Cancer Spectrum 1988, 80, 14–20. [Google Scholar] [CrossRef]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263. [Google Scholar] [CrossRef] [PubMed]
- Blekherman, G.; Laubenbacher, R.; Cortes, D.F.; Mendes, P.; Torti, F.M.; Akman, S.; Torti, S.V.; Shulaev, V. Bioinformatics tools for cancer metabolomics. Metabolomics 2011, 7, 329–343. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food & Drug Administration. U.S. Department of Health and Human Services. 2017. Available online: https://www.fda.gov/ (accessed on 30 August 2018).
- Farshidfar, F.; Weljie, A.M.; Kopciuk, K.; Buie, W.D.; MacLean, A.; Dixon, E.; Sutherland, F.R.; Molckovsky, A.; Vogel, H.J.; Bathe, O.F. Serum metabolomic profile as a means to distinguish stage of colorectal cancer. Genome Med. 2012, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Farshidfar, F.; Weljie, A.M.; Kopciuk, K.A.; Hilsden, R.; McGregor, S.E.; Buie, W.D.; MacLean, A.; Vogel, H.J.; Bathe, O.F. A validated metabolomic signature for colorectal cancer: Exploration of the clinical value of metabolomics. Br. J. Cancer 2016, 115, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Katz, R. Biomarkers and surrogate markers: An fda perspective. NeuroRx 2004, 1, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Krumsiek, J.; Mittelstrass, K.; Do, K.T.; Stückler, F.; Ried, J.; Adamski, J.; Peters, A.; Illig, T.; Kronenberg, F.; Friedrich, N.; et al. Gender-specific pathway differences in the human serum metabolome. Metabolomics 2015, 11, 1815–1833. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Q.; Li, T.; Sun, J.; Liu, X.; Ren, J.; Fei, J. Metabolomic analysis of normal (c57bl/6j, 129s1/svimj) mice by gas chromatography-mass spectrometry: Detection of strain and gender differences. Talanta 2011, 85, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jung, Y.; Park, J.Y.; Lee, S.-H.; Ryu, H.; Hwang, G.-S.; Hwang, G.S. LC/MS-based polar metabolite profiling reveals gender differences in serum from patients with myocardial infarction. J. Pharm. Biomed. Anal. 2015, 115, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Maekawa, K.; Saito, K.; Senoo, Y.; Urata, M.; Murayama, M.; Tajima, Y.; Kumagai, Y.; Saito, Y. Plasma and serum lipidomics of healthy white adults shows characteristic profiles by subjects’ gender and age. PLoS ONE 2014, 9, e91806. [Google Scholar] [CrossRef] [PubMed]
- Holman, R.T.; Smythe, L.; Johnson, S. Effect of sex and age on fatty acid composition of human serum lipids. Am. J. Clin. Nutr. 1979, 32, 2390–2399. [Google Scholar] [CrossRef] [PubMed]
- Putri, S.P.; Yamamoto, S.; Tsugawa, H.; Fukusaki, E. Current metabolomics: Technological advances. J. Biosci. Bioeng. 2013, 116, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Broadhurst, D.I.; Atherton, H.J.; Goodacre, R.; Griffin, J.L. Systems level studies of mammalian metabolomes: The roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem. Soc. Rev. 2011, 40, 387–426. [Google Scholar] [CrossRef] [PubMed]
- Kvitvang, H.F.N.; Kristiansen, K.A.; Lien, S.K.; Bruheim, P. Mass Spectrometry in Metabolomics: Methods and Protocols; Humana Press: New York, NY, USA, 2014; pp. 137–145. [Google Scholar]
- Emwas, A.H.M.; Salek, R.M.; Griffin, J.L.; Merzaban, J. Nmr-based metabolomics in human disease diagnosis: Applications, limitations, and recommendations. Metabolomics 2013, 9, 1048–1072. [Google Scholar] [CrossRef]
- Qiu, Y.; Reed, D. Gas Chromatography in Metabolomics Study; IntechOpen: London, UK, 2014; pp. 83–100. [Google Scholar]
- Lind, M.V.; Savolainen, O.I.; Ross, A.B. The use of mass spectrometry for analysing metabolite biomarkers in epidemiology: Methodological and statistical considerations for application to large numbers of biological samples. Eur. J. Epidemiol. 2016, 31, 717–733. [Google Scholar] [CrossRef] [PubMed]
- West-Nielsen, M.; Høgdall, E.V.; Marchiori, E.; Høgdall, C.K.; Schou, C.; Heegaard, N.H.H. Sample handling for mass spectrometric proteomic investigations of human sera. Anal. Chem. 2005, 77, 5114–5123. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.J.; Gelfand, C.A.; Haywood, B.C.; Warunek, D.J.; Yi, J.; Schuchard, M.D.; Mehigh, R.J.; Cockrill, S.L.; Scott, G.B.I.; Tammen, H.; et al. Hupo plasma proteome project specimen collection and handling: Towards the standardization of parameters for plasma proteome samples. Proteomics 2005, 5, 3262–3277. [Google Scholar] [CrossRef] [PubMed]
- Anton, G.; Wilson, R.; Yu, Z.-H.; Prehn, C.; Zukunft, S.; Adamski, J.; Heier, M.; Meisinger, C.; Römisch-Margl, W.; Wang-Sattler, R.; et al. Pre-analytical sample quality: Metabolite ratios as an intrinsic marker for prolonged room temperature exposure of serum samples. PLoS ONE 2015, 10, e0121495. [Google Scholar] [CrossRef] [PubMed]
- Breier, M.; Wahl, S.; Prehn, C.; Fugmann, M.; Ferrari, U.; Weise, M.; Banning, F.; Seissler, J.; Grallert, H.; Adamski, J.; et al. Targeted metabolomics identifies reliable and stable metabolites in human serum and plasma samples. PLoS ONE 2014, 9, e89728. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, F.; Lobentam, E.-M.; Konig, P.; Utermann, G.; Dieplinger, H. Effect of sample storage on the measurement of lipoprotein[a], apolipoproteins b and a-iv, total and high density lipoprotein cholesterol and trigiycerides. J. Lipid Res. 1994, 35, 1318–1328. [Google Scholar] [PubMed]
- Zivkovic, A.M.; Wiest, M.M.; Nguyen, U.T.; Davis, R.; Watkins, S.M.; German, J.B. Effects of sample handling and storage on quantitative lipid analysis in human serum. Metabolomics 2009, 5, 507. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Peter, A.; Franken, H.; Zhao, X.; Neukamm, S.S.; Rosenbaum, L.; Lucio, M.; Zell, A.; Häring, H.U.; Xu, G.; et al. Preanalytical aspects and sample quality assessment in metabolomics studies of human blood. Clin. Chem. 2013, 59, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Phinney, K.W.; Ballihaut, G.; Bedner, M.; Benford, B.S.; Camara, J.E.; Christopher, S.J.; Davis, W.C.; Dodder, N.G.; Eppe, G.; Lang, B.E.; et al. Development of a standard reference material for metabolomics research. Anal. Chem. 2013, 85, 11732–11738. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, S.E.; Moll, H.A.; Steyerberg, E.W.; Donders, A.R.T.; Derksen-Lubsen, G.; Grobbee, D.E.; Moons, K.G.M. External validation is necessary in prediction research. J. Clin. Epidemiol. 2003, 56, 826–832. [Google Scholar] [CrossRef]
- Usher-Smith, J.A.; Harshfield, A.; Saunders, C.L.; Sharp, S.J.; Emery, J.; Walter, F.M.; Muir, K.; Griffin, S.J. External validation of risk prediction models for incident colorectal cancer using uk biobank. Br. J. Cancer 2018, 118, 750. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Health and Human Services, National Institute of Health. The Early Detection Research Network. 2011. Available online: https://edrn.nci.nih.gov/docs/EDRN5.pdf (accessed on 31 December 2011).
- Pepe, M.S.; Etzioni, R.; Feng, Z.; Potter, J.D.; Thompson, M.L.; Thornquist, M.; Winget, M.; Yasui, Y. Phases of biomarker development for early detection of cancer. J. Natl. Cancer Inst. 2001, 93, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Bowling, F.G.; Thomas, M. Analyzing the metabolome. In Clinical Bioinformatics; Trent, R., Ed.; Springer: New York, NY, USA, 2014; pp. 31–45. [Google Scholar]
- Gu, H.; Chen, H.; Pan, Z.; Jackson, A.U.; Talaty, N.; Xi, B.; Kissinger, C.; Duda, C.; Mann, D.; Raftery, D.; et al. Monitoring diet effects via biofluids and their implications for metabolomics studies. Anal. Chem. 2007, 79, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.W.M. The Handbook of Metabolomics; Springer: New York, NY, USA, 2012; pp. 7–27. [Google Scholar]
- Fliniaux, O.; Gaillard, G.; Lion, A.; Cailleu, D.; Mesnard, F.; Betsou, F. Influence of common preanalytical variations on the metabolic profile of serum samples in biobanks. J. Biomol. NMR 2011, 51, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W. Good laboratory practice in analytical laboratory. J. Sci. 2005, 1, 93–94. [Google Scholar]
- World Health Organization. Handbook: Good Laboratory Practice (glp): Quality Practices for Regulated Non-Clinical Research and Development—2nd ed. 2009. Available online: http://apps.who.int/medicinedocs/en/d/Js19719en/ (accessed on 30 August 2018).
- Khleif, S.N.; Doroshow, J.H.; Hait, W.N. Aacr-fda-nci cancer biomarkers collaborative consensus report: Advancing the use of biomarkers in cancer drug development. Clin. Cancer Res. 2010, 16, 3299–3318. [Google Scholar] [CrossRef] [PubMed]
- Nordström, A.; Lewensohn, R. Metabolomics: Moving to the clinic. J. Neuroimmune Pharmacol. 2010, 5, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.-K.; Lord, G. Current developments in lc-ms for pharmaceutical analysis. Biol. Pharm. Bull. 2002, 25, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Nordström, A.; O’Maille, G.; Qin, C.; Siuzdak, G. Nonlinear data alignment for UPLC-MS and HPLC-MS based metabolomics: Quantitative analysis of endogenous and exogenous metabolites in human serum. Anal. Chem. 2006, 78, 3289–3295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, L.; Shen, J.; Cao, B.; Cheng, T.; Zhao, T.; Liu, X.; Zhang, H. Metabolic signatures of esophageal cancer: Nmr-based metabolomics and uhplc-based focused metabolomics of blood serum. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.C.; Kruppa, G.; Dasseux, J.L. Metabolomics applications of FT-ICR mass spectrometry. Mass Spectrom. Rev. 2005, 24, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Ghaste, M.; Mistrik, R.; Shulaev, V. Applications of fourier transform ion cyclotron resonance (ft-icr) and orbitrap based high resolution mass spectrometry in metabolomics and lipidomics. Int. J. Mol. Sci. 2016, 17, 816. [Google Scholar] [CrossRef] [PubMed]
- Alder, L.; Greulich, K.; Kempe, G.; Vieth, B. Residue analysis of 500 high priority pesticides: Better by gc-ms or lc-ms/ms? Mass Spectrom. Rev. 2006, 25, 838–865. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, W.; Gergov, M.; Goerner, M. Ms/ms-libraries with triple quadrupole-tandem mass spectrometers for drug identification and drug screening. Analusis 2000, 28, 934–941. [Google Scholar] [CrossRef]
- Wu, A.H.; French, D. Implementation of liquid chromatography/mass spectrometry into the clinical laboratory. Clin. Chim. Acta 2013, 420, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Phillips, K.A.; Van Bebber, S.; Issa, A.M. Diagnostics and biomarker development: Priming the pipeline. Nat. Rev. Drug Discov. 2006, 5, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Hajian-Tilaki, K. Sample size estimation in diagnostic test studies of biomedical informatics. J. Biomed. Inform. 2014, 48, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, D.F. How to improve reliability and efficiency of research about molecular markers: Roles of phases, guidelines, and study design. J. Clin. Epidemiol. 2007, 60, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- The COSMOS Project. Cosmo—Coordination of Standards in Metabolomics. Available online: http://www.cosmos-fp7.eu/ (accessed on 12 December 2016).
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Supreme court decision. Mayo Collaborative Services v. Prometheus Laboratories, Inc. No. 10-1150, Court, U.S.S., Ed. 2012. Available online: https://www.supremecourt.gov/opinions/11pdf/10-1150.pdf (accessed on 12 December 2016).
- Woodcock, J.; Woosley, R. The FDA critical path initiative and its influence on new drug development. Ann. Rev. Med. 2008, 59, 1–12. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food & Drug Administration. Challenges and Opportunities Report—March 2004. U.S. Department of Health and Human Services, 2004. Available online: http://www.who.int/intellectualproperty/documents/en/FDAproposals.pdf (accessed on 12 December 2016).
- United States Office of the Federal Register. Code of Federal Regulations Title 42. U.S. Food and Drug Administration, 2016. Available online: https://en.wikipedia.org/wiki/Title_42_of_the_Code_of_Federal_Regulations (accessed on 21 July 2016).
- National Human Genome Research Institute. Regulation of Genetic Testing. Available online: https://www.genome.gov/10002335/regulation-of-genetic-tests/-al-5 (accessed on 12 December 2016).
- U.S. Food & Drug Administration. Draft Guidance for Industry, Food and Drug Administration Staff, and Clinical Laboratories: Fda Notification and Medical Device Reporting for Laboratory Developed Tests (ldts). U.S. Department of Health and Human Services, 2014. Available online: https://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm416684.pdf (accessed on 3 October 2014).
- Sager, M.; Chien Yeat, N.; Pajaro-Van der Stadt, S.; Lin, C.; Ren, Q.; Lin, J. Transcriptomics in cancer diagnostics: Developments in technology, clinical research and commercialization. Expert Rev. Mol. Diagn. 2015, 15, 1589–1603. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Important Factors for Consideration | Conditions and Explanation | Reference Study |
|---|---|---|
| Experimental Conditions | ||
| Choice of methodology (technology) | Choice of instrumentation should take into consideration the class of metabolites and biological functions of interest. | Putri et al. [18] |
| Analysis batches should be balanced with comparator groups, taking into account potential confounders. Corrections must be made for batch variation. | Dunn et al. [19] | |
| Sample preparation | Chemical derivatizations made during the preparation procedure should be designed to specifically react with the target chemical structure and cause minimum alterations to the unintended sites. | Kvitvang et al. [20] |
| Patient Factors | ||
| Diet and drug interactions | Diet and drug exposure may affect metabolite quantities. Therefore, ideally, blood samples should be taken from the patient after 8–12 h of fasting. | Emwas et al. [21] |
| Physical activity | To avoid the metabolite fluctuation as a result of physical activity, no physical exercise should be done before sample extraction, unless that is a component of the biomarker. | Emwas et al. [21] |
| Age | Disease state and controls should be age matched. | Ishikawa et al. [16] |
| Sex | Sex composition of study cohorts should be considered when designing a balanced and unbiased study. | Krumsiek et al. [13] |
| Sample Processing | ||
| Processing time | Tissue dissection should be carried out immediately to minimize sample degradation. Sample collection and processing standardized operating procedures (SOPs) are essential. | Emwas et al. [21] |
| Processing reagents | The type of coagulant used to create serum samples from plasma might have an effect on the ionization process in analysis. | Qiu et al. [22] Lind et al. [23] |
| Sample storage and preservation | Samples should be stored at –80 °C to minimize changes in metabolite concentrations. | Emwas et al. [21] |
| While some metabolites may be altered at –20 °C, valid metabolomic signatures can be derived as long as comparator groups are stored at the same temperature. | West- Nielson et al. [24] Rai et al. [25] | |
| Keeping samples on cool packs during processing will reduce degradation as compared to processing at room temperature. | Anton et al. [26] | |
| Time from sample collection to freezing should be minimized to prevent degradation of some classes of metabolites, including amino acids and biogenic amines. | Breier et al. [27] | |
| Processing samples shortly after collection is optimal. Samples from different years of collection will have different metabolomic profiles, and this will need to be considered in the batch design as well as the final analysis. | Lind et al. [23] | |
| Sample Storage | ||
| Freeze/Thaw cycles | Minimizing freeze/thaw cycles will result in less compositional changes. 1–4 freeze/thaw cycles cause little variation but 5+ cycles cause significant variation. | Anton et al. [26] Kronenberg et al. [28] Zivkovic et al. [29] Yin et al. [30] |
| Quality Assurance | ||
| Quality Control samples | Quality Control (QC) samples are analyzed at the same time as unknown samples to minimize standard error (SE) by accounting for biological or analytical variations. | Phinney et al. [31] |
| Internal standards | Internal Standards (IS) are used to validate the identity and to quantify metabolites, and should be added to each sample at the beginning of the preparation process. | Phinney et al. [31] Lind et al. [23] |
| Type of Analytical Platform | Advantages | Disadvantages | References |
|---|---|---|---|
| Nuclear Magnetic Resonance (NMR) | Non-destructive (samples can be recovered for further analysis). Relatively easy sample preparation. Good for complex mixtures (respective intensities of metabolites are not affected by others). NMR Spectra can easily be validated with 2D Spectroscopy methods (TOCSY or STOCSY). | Higher detection threshold than Mass Spectrometry. Requires a large, expensive, and specialized facility (impractical for most clinical labs). | Putri et al. [18] |
| Mass Spectrometry | |||
| Gas Chromatography-Mass Spectrometry (GC-MS) | Detects low molecular weight compounds (e.g., amino acids, sugars, etc.). Compact machine (smaller footprint for clinical laboratory). High peak capacity to cover a wide range of concentrations. Utilizes Electron Ionization (EI) to increase ionization efficiency. Normalized retention indices for each compound. Reliable standard operating procedures (SOPs). | Samples must be volatile in order to pass through the machine (requires compound derivatization). Derivatization alters compounds’ chemical structure which could misrepresent desired compound after fragmentation in MS. Higher molecular weight compounds are insufficiently volatile. | Putri et al. [18] Nordström et al. [43] |
| Liquid Chromatography–Mass Spectrometry (LC-MS) | Analyzes a wide range of metabolites of varying molecular weight. Detects both hydrophilic and hydrophobic compounds. Does not require derivatization. Better compound resolution than GC-MS. | LC-MS can be completed in a few minutes using short columns; but this can create suppression effects. These effects can also be caused by flow injection analysis (FIA) | Putri et al. [18] Lim et al. [44] |
| Ultra-High Performance Liquid Chromatography (UHPLC) | Higher signal-to-noise ratio, sensitivity, and specificity than NMR. Dependent on the detection device that is used (such as MS) | Fractionation requires greater pressure through columns of smaller particle sizes. | Nordström et al. [45] Zhang et al. [46] |
| Fourier-Transform Ion Cyclotron Resonance (FT-ICR) | Lower limit of detection and precise measurement. Simple sample preparation. Most sensitive method for detecting metabolites in complex mixtures. Can use either Electrospray ionization (ESI )or ApCl ionization. | Slower acquisition rates of the Fourier transform mass spectrometry (FTMS). Instrumentation is large and expensive. | Brown et al. [47] Ghaste et al. [48] Lim et al. [44] |
| Triple-Quadrupole Tandem MS (MS/MS) | Can be combined with either GC or LC. Reduces background noise stemming from solvent. Can be combined with Selection Reaction Monitoring (SRM) to further fragment ions and create a more sensitive platform. | Requires precursor ion selection before experiment; single-quadrupole mode must be used before triple-quadrupole. Total number of ions entering ion-trap is restricted, causes difficulty with small amounts of analyte in question (“ion suppression”) | Alder et al. [49] Weinmann et al. [50] Wu et al. [51] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchand, C.R.; Farshidfar, F.; Rattner, J.; Bathe, O.F. A Framework for Development of Useful Metabolomic Biomarkers and Their Effective Knowledge Translation. Metabolites 2018, 8, 59. https://doi.org/10.3390/metabo8040059
Marchand CR, Farshidfar F, Rattner J, Bathe OF. A Framework for Development of Useful Metabolomic Biomarkers and Their Effective Knowledge Translation. Metabolites. 2018; 8(4):59. https://doi.org/10.3390/metabo8040059
Chicago/Turabian StyleMarchand, Calena R., Farshad Farshidfar, Jodi Rattner, and Oliver F. Bathe. 2018. "A Framework for Development of Useful Metabolomic Biomarkers and Their Effective Knowledge Translation" Metabolites 8, no. 4: 59. https://doi.org/10.3390/metabo8040059
APA StyleMarchand, C. R., Farshidfar, F., Rattner, J., & Bathe, O. F. (2018). A Framework for Development of Useful Metabolomic Biomarkers and Their Effective Knowledge Translation. Metabolites, 8(4), 59. https://doi.org/10.3390/metabo8040059