Mind the Gap: Mapping Mass Spectral Databases in Genome-Scale Metabolic Networks Reveals Poorly Covered Areas

 , ,
, ,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Material and Methods
2.1. Chemical Library
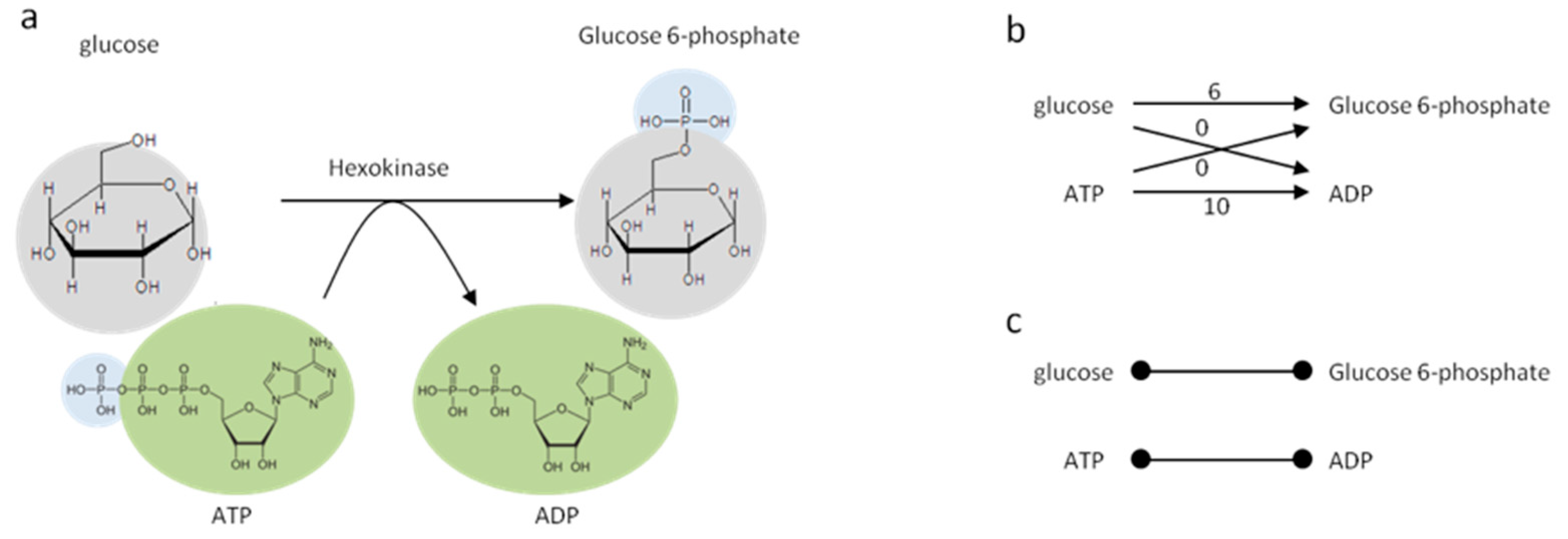
2.2. Human Metabolic Network and Graph Construction
2.3. Network Topology Analysis
2.4. Publication Mapping
3. Results
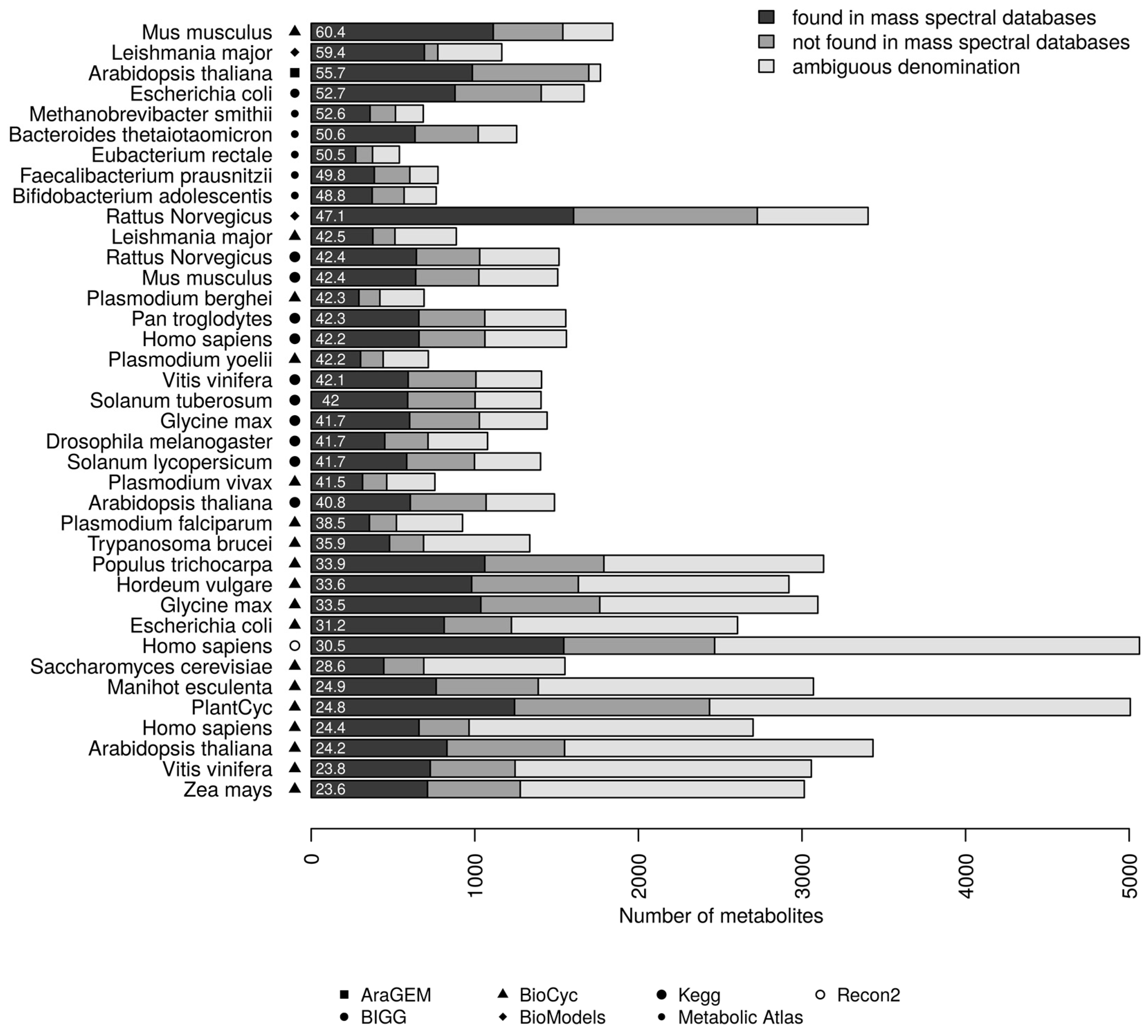
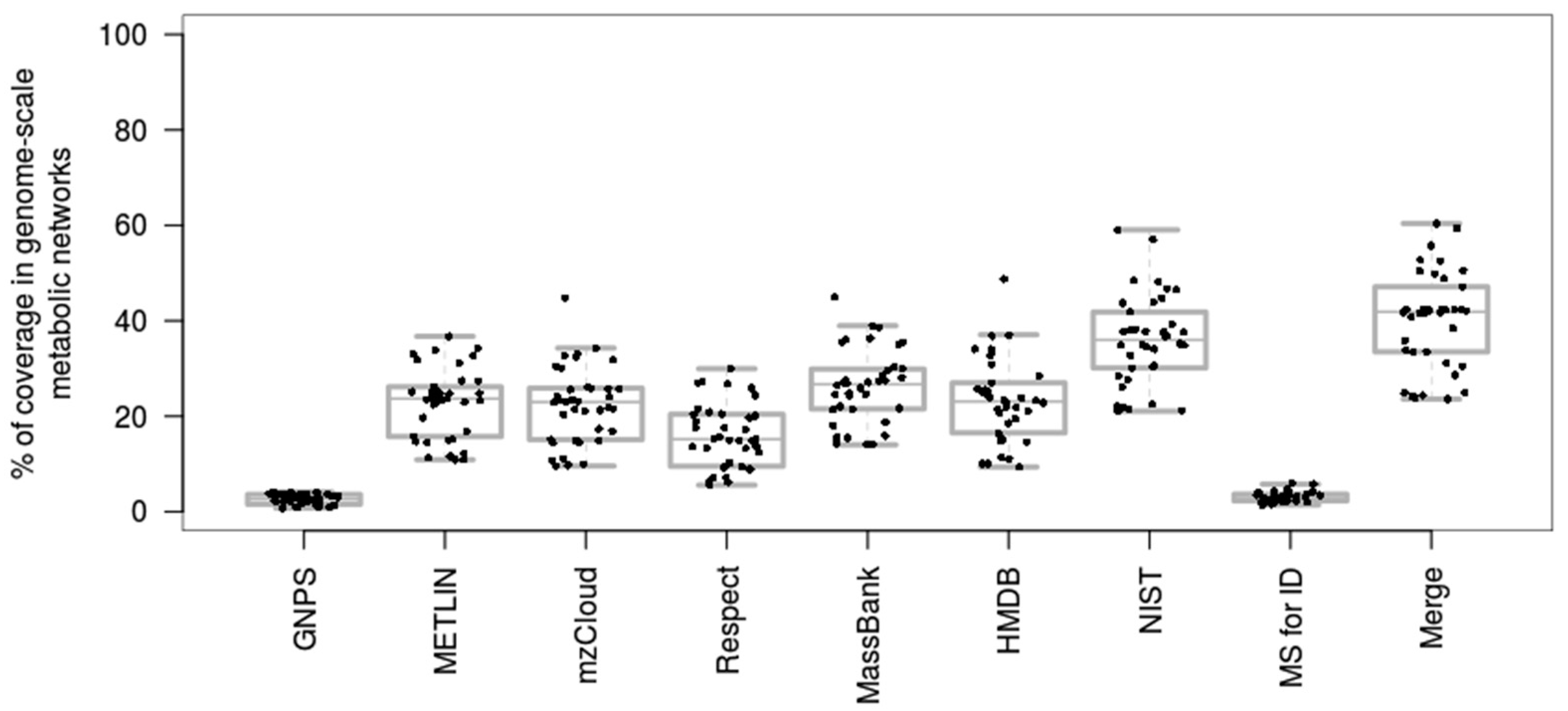
3.1. Coverage of Genome-Scale Metabolic Networks by Mass Spectral Libraries
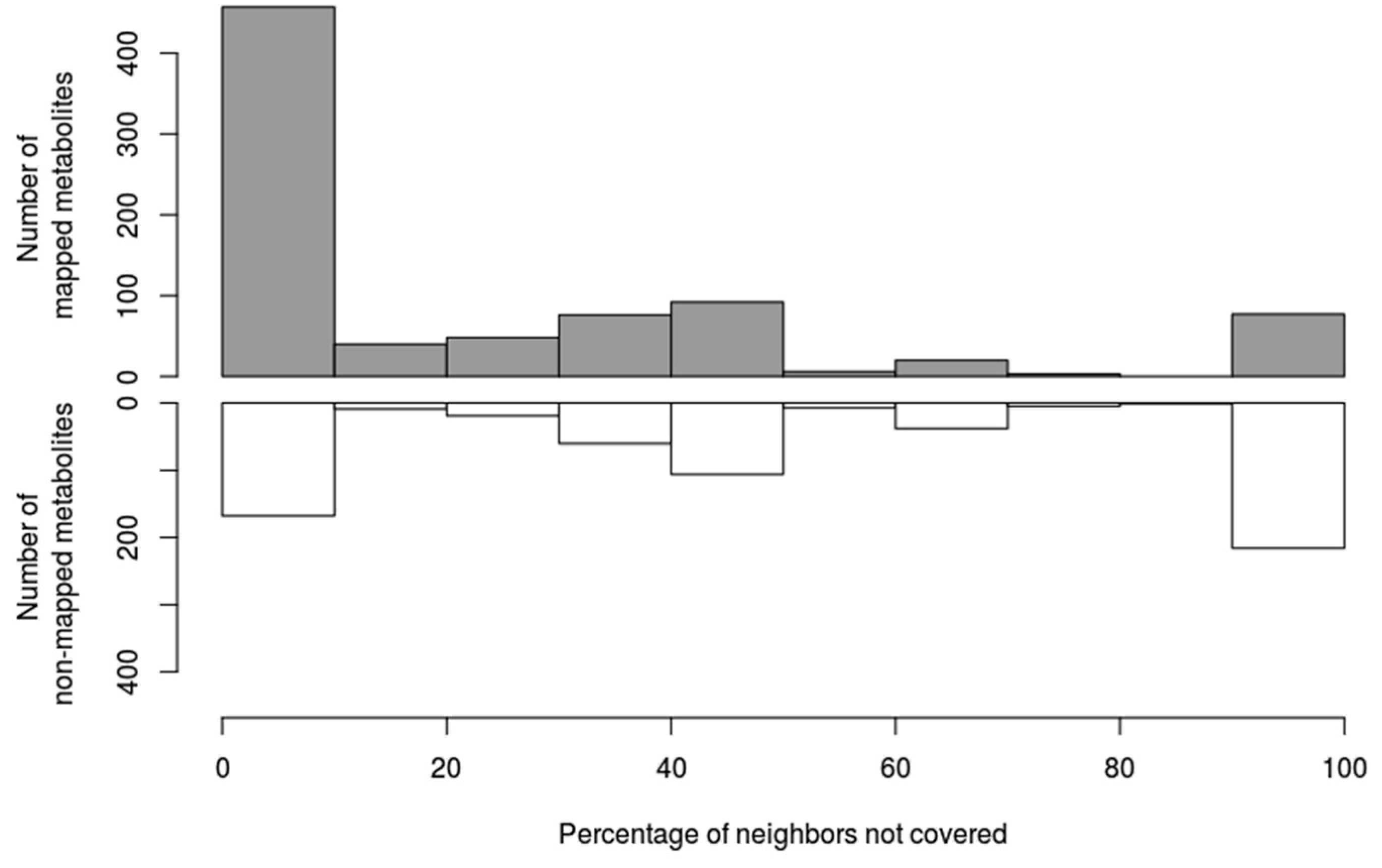
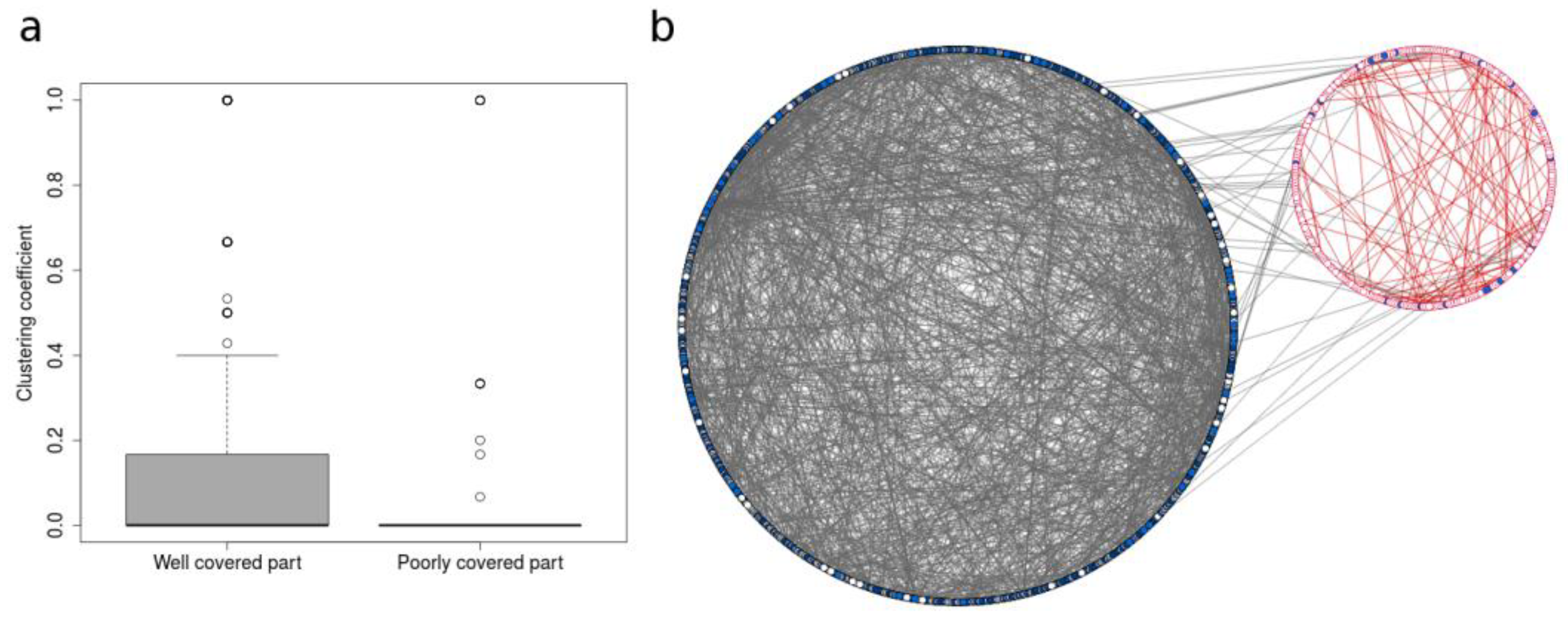
3.2. Deciphering Poorly Covered Parts of the Human Metabolic Network
3.3. Filling Gaps in Poorly Covered Areas of Human Metabolism
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263. [Google Scholar] [CrossRef] [PubMed]
- Panopoulos, A.D.; Yanes, O.; Ruiz, S.; Kida, Y.S.; Diep, D.; Tautenhahn, R.; Herrerías, A.; Batchelder, E.M.; Plongthongkum, N.; Lutz, M.; et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012, 22, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Slebe, F.; Rojo, F.; Vinaixa, M.; García-Rocha, M.; Testoni, G.; Guiu, M.; Planet, E.; Samino, S.; Arenas, E.J.; Beltran, A.; et al. FoxA and LIPG endothelial lipase control the uptake of extracellular lipids for breast cancer growth. Nat. Commun. 2016, 7, 11199. [Google Scholar] [CrossRef] [PubMed]
- Jorge, T.F.; Rodrigues, J.A.; Caldana, C.; Schmidt, R.; van Dongen, J.T.; Thomas-Oates, J.; António, C. Mass spectrometry-based plant metabolomics: Metabolite responses to abiotic stress. Mass Spectrom. Rev. 2016, 35, 620–649. [Google Scholar] [CrossRef] [PubMed]
- Barkal, L.J.; Theberge, A.B.; Guo, C.-J.; Spraker, J.; Rappert, L.; Berthier, J.; Brakke, K.A.; Wang, C.C.C.; Beebe, D.J.; Keller, N.P.; et al. Microbial metabolomics in open microscale platforms. Nat. Commun. 2016, 7, 10610. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Wang, M.; Hyde, E.; da Silva, R.R.; Melnik, A.V.; Protsyuk, I.; Bouslimani, A.; Lim, Y.W.; Wong, R.; Humphrey, G.; et al. Three-Dimensional Microbiome and Metabolome Cartography of a Diseased Human Lung. Cell Host Microbe 2017, 22, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Baran, R.; Brodie, E.L.; Mayberry-Lewis, J.; Hummel, E.; Da Rocha, U.N.; Chakraborty, R.; Bowen, B.P.; Karaoz, U.; Cadillo-Quiroz, H.; Garcia-Pichel, F.; et al. Exometabolite niche partitioning among sympatric soil bacteria. Nat. Commun. 2015, 6, 8289. [Google Scholar] [CrossRef] [PubMed]
- Hollender, J.; Schymanski, E.L.; Singer, H.P.; Ferguson, P.L. Nontarget Screening with High Resolution Mass Spectrometry in the Environment: Ready to Go? Environ. Sci. Technol. 2017, 51, 11505–11512. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Reig, M.; Jaumot, J.; Piña, B.; Moyano, E.; Galceran, M.T.; Tauler, R. Metabolomic analysis of the effects of cadmium and copper treatment in Oryza sativa L. using untargeted liquid chromatography coupled to high resolution mass spectrometry and all-ion fragmentation. Metallomics 2017, 9, 660–675. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Tsugawa, H.; Cajka, T.; Ma, Y.; Lai, Z.; Mehta, S.S.; Wohlgemuth, G.; Barupal, D.K.; Showalter, M.R.; Arita, M.; et al. Identification of small molecules using accurate mass MS/MS search. Mass Spectrom. Rev. 2017, 37, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.-J.; Schultz, A.W.; Wang, J.; Johnson, C.H.; Yannone, S.M.; Patti, G.J.; Siuzdak, G. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat. Protoc. 2013, 8, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2017, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Vinaixa, M.; Schymanski, E.L.; Neumann, S.; Navarro, M.; Salek, R.M.; Yanes, O. Mass spectral databases for LC/MS- and GC/MS-based metabolomics: State of the field and future prospects. TrAC-Trends Anal. Chem. 2016, 78, 23–35. [Google Scholar] [CrossRef]
- Henry, C.S.; DeJongh, M.; Best, A.A.; Frybarger, P.M.; Linsay, B.; Stevens, R.L. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 2010, 28, 977. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, D.; Palsson, B.Ø.; Feist, A.M. Basic and applied uses of genome-scale metabolic network reconstructions of Escherichia coli. Mol. Syst. Biol. 2013, 9, 661. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2015, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Pence, H.E.; Williams, A. ChemSpider: An Online Chemical Information Resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Smith, C.A.; Maille, G.O.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A Metabolite Mass Spectral Database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef]
- Sawada, Y.; Nakabayashi, R.; Yamada, Y.; Suzuki, M.; Sato, M.; Sakata, A.; Akiyama, K.; Sakurai, T.; Matsuda, F.; Aoki, T.; et al. RIKEN tandem mass spectral database (ReSpect) for phytochemicals: A plant-specific MS/MS-based data resource and database. Phytochemistry 2012, 82, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828. [Google Scholar] [CrossRef] [PubMed]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Swainston, N.; Fleming, R.M.T.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Haraldsdóttir, H.S.; Thiele, I.; Fleming, R.M. Comparative evaluation of open source software for mapping between metabolite identifiers in metabolic network reconstructions: Application to Recon 2. J. Cheminform. 2014, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–W633. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef] [PubMed]
- Degtyarenko, K.; de Matos, P.; Ennis, M.; Hastings, J.; Zbinden, M.; McNaught, A.; Alcántara, R.; Darsow, M.; Guedj, M.; Ashburner, M. ChEBI: A database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2008, 36, D344–D350. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, U.N.; Albert, R.; Kumara, S. Near linear time algorithm to detect community structures in large-scale networks. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2007, 76, 036106. [Google Scholar] [CrossRef] [PubMed]
- Frainay, C.; Jourdan, F. Computational methods to identify metabolic sub-networks based on metabolomic profiles. Brief. Bioinform. 2017, 18, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Bryant, S.H. PUG-SOAP and PUG-REST: Web services for programmatic access to chemical information in PubChem. Nucleic Acids Res. 2015, 43, W605–W611. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Mogas, A.; Sales-Pardo, M.; Navarro, M.; Guimerà, R.; Yanes, O. iMet: A network-based computational tool to assist in the annotation of metabolites from tandem mass spectra. Anal. Chem. 2017, 89, 3474–3482. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Pon, A.; Wilson, M.; Greiner, R.; Wishart, D. CFM-ID: A web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014, 42, W94–W99. [Google Scholar] [CrossRef] [PubMed]
- Dührkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Böcker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, G.; Haldiya, P.K.; Willighagen, E.; Kind, T.; Fiehn, O. The Chemical Translation Service—A web-based tool to improve standardization of metabolomic reports. Bioinformatics 2010, 26, 2647–2648. [Google Scholar] [CrossRef] [PubMed]
- Schymanski, E.L.; Williams, A.J. Open Science for Identifying “Known Unknown” Chemicals. Environ. Sci. Technol. 2017, 51, 5357–5359. [Google Scholar] [CrossRef] [PubMed]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Dräger, A.; Mih, N.; Gatto, F.; Nilsson, A.; Preciat-Gonzalez, G.A.; Aurich, M.K.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name (from Network) | PubChem CID | InChIKey |
|---|---|---|
| (25R)-3alpha,7alpha,12alpha-trihydroxy-5beta-cholestan-26-oyl-CoA(4-) | 15942889 | MNYDLIUNNOCPHG-FJWDCHQMSA-N |
| 12-oxo-c-LTB3 | 122164853 | ZFHPYBQKHVEFHO-LECUDPRGSA-N |
| 3alpha,7alpha,12alpha-Trihydroxy-5beta-cholestanoate | 440460 | CNWPIIOQKZNXBB-SQZFNYHNSA-N |
| 3alpha,7alpha,12alpha-trihydroxy-5beta-cholestan-26-al | 193321 | XJZGNVBLVFOSKJ-XZULNKEGSA-N |
| 12-oxo-leukotriene B4 | 5280876 | SJVWVCVZWMJXOK-NOJHDUNKSA-N |
| 20-CoA-20-oxo-leukotriene B4 | 53481505 | WLWKYZHFLKRKEU-WCOJVGLOSA-J |
| 5beta-cholestane-3alpha,7alpha,12alpha,26-tetrol | 439479 | USFJGINJGUIFSY-XZULNKEGSA-N |
| (4R,5S)-4,5,6-trihydroxy-2,3-dioxohexanoate | 440390 | GJQWCDSAOUMKSE-STHAYSLISA-N |
| 20-carboxy-leukotriene-B4 | 5280877 | SXWGPVJGNOLNHT-VFLUTPEKSA-N |
| 5beta-cholestane-3alpha,7alpha,12alpha-triol | 160520 | RIVQQZVHIVNQFH-XJZYBRFWSA-N |
| 3-oxo-tetracosa-12,15,18,21-all-cis-tetraenoyl-CoA | 131769900 | HPMVBGKWFWCZAY-JDTXFHFDSA-N |
| 6-pyruvoyl-5,6,7,8-tetrahydropterin | 128973 | WBJZXBUVECZHCE-UHFFFAOYSA-N |
| Hydroxymethylbilane | 788 | WDFJYRZCZIUBPR-UHFFFAOYSA-N |
| 5beta-cholestane-3alpha,7alpha,12alpha,25-tetrol | 160520 | RIVQQZVHIVNQFH-XJZYBRFWSA-N |
| 3(S)-hydroxy-tetracosa-12,15,18,21-all-cis-tetraenoyl-CoA | 53477712 | NTIXPPFPXLYJCT-OWOWEXKPSA-N |
| Uroporphyrinogen III | 1179 | HUHWZXWWOFSFKF-UHFFFAOYSA-N |
| 12-oxo-20-hydroxy-leukotriene B4 | 53481459 | CZWPUWRHQBAXJS-PABROBRYSA-N |
| 3-oxo-all-cis-6,9,12,15,18-tetracosapentaenoyl-CoA | 131769894 | UQPANOGFYCZRAV-UWOIJHEUSA-N |
| all-cis-10,13,16,19-docosatetraenoyl-CoA | 71627222 | BEEQBBPNTYBGDP-BUSXXEPMSA-J |
| kinetensin | 53481569 | PANUJGMSOSQAAY-HAGIGRARSA-N |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frainay, C.; Schymanski, E.L.; Neumann, S.; Merlet, B.; Salek, R.M.; Jourdan, F.; Yanes, O. Mind the Gap: Mapping Mass Spectral Databases in Genome-Scale Metabolic Networks Reveals Poorly Covered Areas. Metabolites 2018, 8, 51. https://doi.org/10.3390/metabo8030051
Frainay C, Schymanski EL, Neumann S, Merlet B, Salek RM, Jourdan F, Yanes O. Mind the Gap: Mapping Mass Spectral Databases in Genome-Scale Metabolic Networks Reveals Poorly Covered Areas. Metabolites. 2018; 8(3):51. https://doi.org/10.3390/metabo8030051
Chicago/Turabian StyleFrainay, Clément, Emma L. Schymanski, Steffen Neumann, Benjamin Merlet, Reza M. Salek, Fabien Jourdan, and Oscar Yanes. 2018. "Mind the Gap: Mapping Mass Spectral Databases in Genome-Scale Metabolic Networks Reveals Poorly Covered Areas" Metabolites 8, no. 3: 51. https://doi.org/10.3390/metabo8030051
APA StyleFrainay, C., Schymanski, E. L., Neumann, S., Merlet, B., Salek, R. M., Jourdan, F., & Yanes, O. (2018). Mind the Gap: Mapping Mass Spectral Databases in Genome-Scale Metabolic Networks Reveals Poorly Covered Areas. Metabolites, 8(3), 51. https://doi.org/10.3390/metabo8030051