The Energetic Viability of Δ1-Piperideine Dimerization in Lysine-derived Alkaloid Biosynthesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Result
2.1. Benchmark Calculations
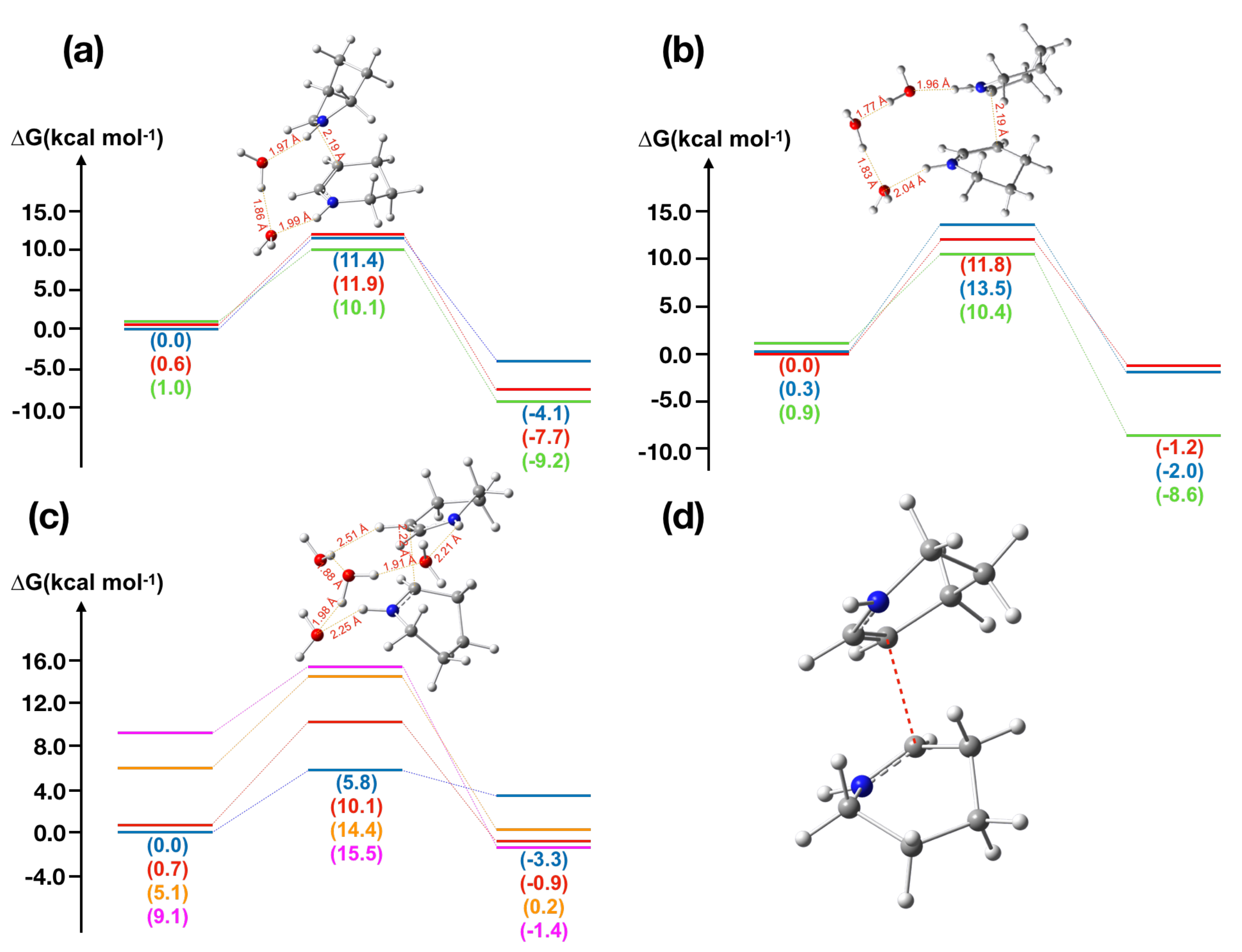
2.2. -Piperideine Dimerization
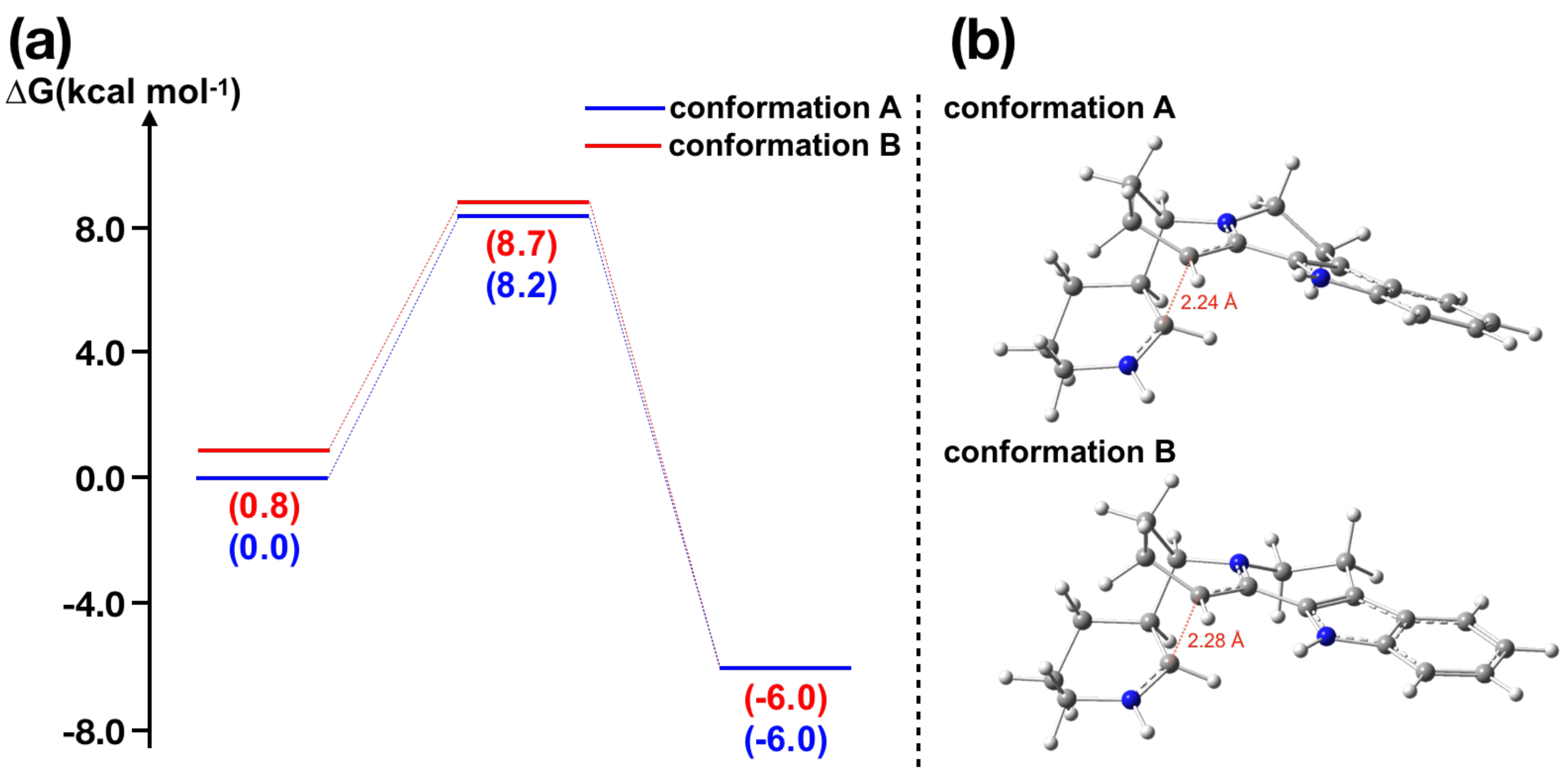
2.3. Nitramidine Biosynthesis
3. Discussion
4. Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| QA | quinolizidine alkaloid |
| LDC | lysine decarboxylase |
| THS | thebaine synthase |
| CAO | copper amine oxidase |
| DFT | density functional theory |
References
- Rai, A.; Saito, K.; Yamazaki, M. Integrated omics analysis of specialized metabolism in medicinal plants. Plant J. 2017, 90, 764–787. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Rai, A.; Yoshimoto, N.; Saito, K. Perspective: Functional genomics towards new biotechnology in medicinal plants. Plant Biotechnol. Rep. 2018, 12, 69–75. [Google Scholar] [CrossRef]
- Sato, H.; Wang, C.; Yamazaki, M.; Saito, K.; Uchiyama, M. Computational study on a puzzle in the biosynthetic pathway of anthocyanin: Why is an enzymatic oxidation/ reduction process required for a simple tautomerization? PLoS ONE 2018, 13, e0198944. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Narita, K.; Minami, A.; Yamazaki, M.; Wang, C.; Suemune, H.; Nagano, S.; Tomita, T.; Oikawa, H.; Uchiyama, M. Theoretical Study of Sesterfisherol Biosynthesis: Computational Prediction of Key Amino Acid Residue in Terpene Synthase. Sci. Rep. 2018, 8, 2473. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Patel, A.; Houk, K.N. Transannular [6 + 4] and Ambimodal Cycloaddition in the Biosynthesis of Heronamide A. J. Am. Chem. Soc. 2015, 137, 13518–13523. [Google Scholar] [CrossRef] [PubMed]
- Newmister, S.A.; Li, S.; Garcia-Borràs, M.; Sanders, J.N.; Yang, S.; Lowell, A.N.; Yu, F.; Smith, J.L.; Williams, R.M.; Houk, K.N.; et al. Structural basis of the Cope rearrangement and cyclization in hapalindole biogenesis. Nat. Chem. Biol. 2018, 14, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Tantillo, D.J. Does Nature Know Best? Pericyclic Reactions in the DaphniphyllumAlkaloid-Forming Cation Cascade. Org. Lett. 2016, 18, 4482–4484. [Google Scholar] [CrossRef] [PubMed]
- Bunsupa, S.; Yamazaki, M.; Saito, K. Quinolizidine alkaloid biosynthesis: Recent advances and future prospects. Front. Plant sci. 2012, 3, 239. [Google Scholar] [CrossRef] [PubMed]
- Ohmiya, S.; Saito, K.; Murakoshi, I. Lupine alkaloids. In The Alkaloids: Chemistry and Pharmacology; Cordell, G.A., Ed.; Academic Press: London, UK, 1995; Volume 47, pp. 1–114. [Google Scholar]
- Michael, J.P. Indolizidine and quinolizidine alkaloids. Nat. Prod. Rep. 2008, 25, 139–165. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.N.; Spenser, I.D. Biosynthesis of the piperidine nucleus: The occurrence of the pathways from lysine. J. Biol. Chem. 1970, 9, 2329–2334. [Google Scholar] [CrossRef]
- Leistner, E.; Spenser, I.D. Biosynthesis of the piperidine nucleus. Incorporation of chirally labeled cadaverine-1-3H. J. Am. Chem. Soc. 1973, 95, 4715–4725. [Google Scholar] [CrossRef] [PubMed]
- Golebiewski, W.M.; Spenser, I.D. The biosynthesis of the lupine alkaloids. A reexamination. J. Am. Chem. Soc. 1976, 98, 6726–6728. [Google Scholar] [CrossRef]
- Leeper, F.J.; Grue-Sorensen, G.; Spenser, I.D. Biosynthesis of the quinolizidine alkaloids. Incorporation of Δ1-piperideine into matrine. Can. J. Chem. 1981, 59, 106–115. [Google Scholar] [CrossRef]
- Golebiewski, W.M.; Spenser, I.D. Biosynthesis of the lupine alkaloids. I. Lupinine. Can. J. Chem. 1985, 63, 2707–2718. [Google Scholar] [CrossRef]
- Golebiewski, W.M.; Spenser, I.D. Biosynthesis of the lupine alkaloids. II. Sparteine and Lupanine. Can. J. Chem. 1988, 66, 1734–1748. [Google Scholar] [CrossRef]
- Bunsupa, S.; Katayama, K.; Ikeura, E.; Oikawa, A.; Toyooka, K.; Saito, K.; Yamazaki, M. Lysine Decarboxylase Catalyzes the First Step of Quinolizidine Alkaloid Biosynthesis and Coevolved with Alkaloid Production in Leguminosae. Plant Cell 2012, 24, 1202–1216. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Suzuki, H.; Takamatsu, S.; Murakoshi, I. Acyltransferases for lupin alkaloids in lupinus hirsutus. Phytochemistry 1993, 32, 87–91. [Google Scholar] [CrossRef]
- Suzuki, H.; Koike, Y.; Murakoshi, I.; Saito, K. Subcellular localization of acyltransferases for quinolizidine alkaloid biosynthesis in Lupinus. Phytochemistry 1996, 42, 1557–1562. [Google Scholar] [CrossRef]
- Suzuki, H.; Murakoshi, I.; Saito, K. A novel O-tigloyltransferase for alkaloid biosynthesis in plants. Purification, characterization, and distribution in Lupinus plants. J. Biol. Chem. 1994, 269, 15853–15860. [Google Scholar] [PubMed]
- Okada, T.; Hirai, M.Y.; Suzuki, H.; Yamazaki, M.; Saito, K. Molecular Characterization of a Novel Quinolizidine Alkaloid O-Tigloyltransferase: cDNA Cloning, Catalytic Activity of Recombinant Protein and Expression Analysis in Lupinus Plants. Plant Cell Physiol. 2005, 46, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Nagy, I.; Mancinotti, D.; Otterbach, S.L.; Andersen, T.B.; Motawia, M.S.; Asp, T.; Geu-Flores, F. Transcript profiling of a bitter variety of narrow-leafed lupin to discover alkaloid biosynthetic genes. J. Exp. Bot. 2017, 68, 5527–5537. [Google Scholar] [CrossRef] [PubMed]
- Wanner, J.M.; Koomen, G.-J. Oxidative Deamination of Tetrahydroanabasine with o-Quinones: An Easy Entry to Lupinine, Sparteine, and Anabasine. J. Org. Chem. 1996, 61, 5581–5586. [Google Scholar] [CrossRef]
- Gomm, A.; Lewis, W.; Green, A.P.; O’Reilly, E. A New Generation of Smart Amine Donors for Transaminase-Mediated Biotransformations. Chem. Eur. J. 2016, 22, 12692–12695. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. IV. The role of the exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Boese, A.D.; Martin, J.M.L. Development of density functionals for thermochemical kinetics. J. Chem. Phys. 2004, 121, 3405–3416. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange-Correlation Functional Using the Coulomb- Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Account. 2007, 120, 215–241. [Google Scholar] [CrossRef]
- Perdew, P.J.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward chemical accuracy in the computation of NMR shieldings: The PBE0 model. Chem. Phys. Lett. 1999, 298, 113–119. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta-Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1997, 108, 664–675. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106–16. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Rouchaud, A.; Braekman, J.-C. Synthesis of New Analogues of the Tetraponerines. Eur. J. Org. Chem. 2009, 2009, 2666–2674. [Google Scholar] [CrossRef]
- Tantillo, D.J. Applied Theoretical Organic Chemistry; World Scientific Publishing Co. Pte Ltd.: Singapore, 2018. [Google Scholar] [CrossRef]
- Wanner, M.J.; Koomen, G.-J. Stereoselective Synthesis of the Indole Alkaloids Nitrarine, Nitramidine, and Isomers. A Biomimetic Approach. J. Org. Chem. 1994, 59, 7479–7484. [Google Scholar] [CrossRef]
- Ibragimov, A.A.; Maekh, S. K.; Yunusov, S.Y. The Structure of Nitramidine. Chem. Nat. Compd. 1975, 11, 295–296. [Google Scholar] [CrossRef]
- Gravel, E.; Poupon, E. Biosynthesis and biomimetic synthesis of alkaloids isolated from plants of the Nitraria and Myrioneuron genera: An unusual lysine-based metabolism. Nat. Prod. Rep. 2010, 27, 32–56. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y. Lignans, Lignins, and Resveratrols. In From Biosynthesis to Total Synthesis: Strategies and Tactics for Natural Products; Zografos, A.L., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 333–335. [Google Scholar]
- Chen, X.; Hagel, J.M.; Chang, L.; Tucker, J.E.; Shiigi, S.A.; Yelpaala, Y.; Chen, H.-Y.; Estrada, R.; Colbeck, J.; Enquist-Newman, M.; et al. A pathogenesis-related 10 protein catalyzes the final step in thebaine biosynthesis. Nat. Chem. Biol. 2018, 14, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions-the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Ishida, K.; Morokuma, K.; Komornicki, A. The intrinsic reaction coordinate. An ab initio calculation for HNC→HCN and H− + CH4 → CH4 + H−. J. Chem. Phys. 1977, 66, 2153–2156. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Schlegel, H.B.; Gonzalez, C. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Page, M.; Doubleday, C.; McIver, J.W. Following steepest descent reaction paths. The use of higher energy derivatives with ab initio electronic structure methods. J. Chem. Phys. 1990, 93, 5634–5642. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Mennuci, B.; Tomasi, J. Ab initio study of ionic solutions by a polarizable continuum dielectric model. Chem. Phys. Lett. 1998, 286, 253–260. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. (THEOCHEM) 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Tomasi, J.; Menucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, H.; Uchiyama, M.; Saito, K.; Yamazaki, M. The Energetic Viability of Δ1-Piperideine Dimerization in Lysine-derived Alkaloid Biosynthesis. Metabolites 2018, 8, 48. https://doi.org/10.3390/metabo8030048
Sato H, Uchiyama M, Saito K, Yamazaki M. The Energetic Viability of Δ1-Piperideine Dimerization in Lysine-derived Alkaloid Biosynthesis. Metabolites. 2018; 8(3):48. https://doi.org/10.3390/metabo8030048
Chicago/Turabian StyleSato, Hajime, Masanobu Uchiyama, Kazuki Saito, and Mami Yamazaki. 2018. "The Energetic Viability of Δ1-Piperideine Dimerization in Lysine-derived Alkaloid Biosynthesis" Metabolites 8, no. 3: 48. https://doi.org/10.3390/metabo8030048
APA StyleSato, H., Uchiyama, M., Saito, K., & Yamazaki, M. (2018). The Energetic Viability of Δ1-Piperideine Dimerization in Lysine-derived Alkaloid Biosynthesis. Metabolites, 8(3), 48. https://doi.org/10.3390/metabo8030048