
Development of Database Assisted Structure Identification (DASI) Methods for Nontargeted Metabolomics

Abstract
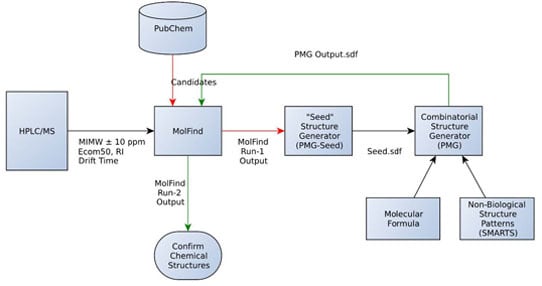
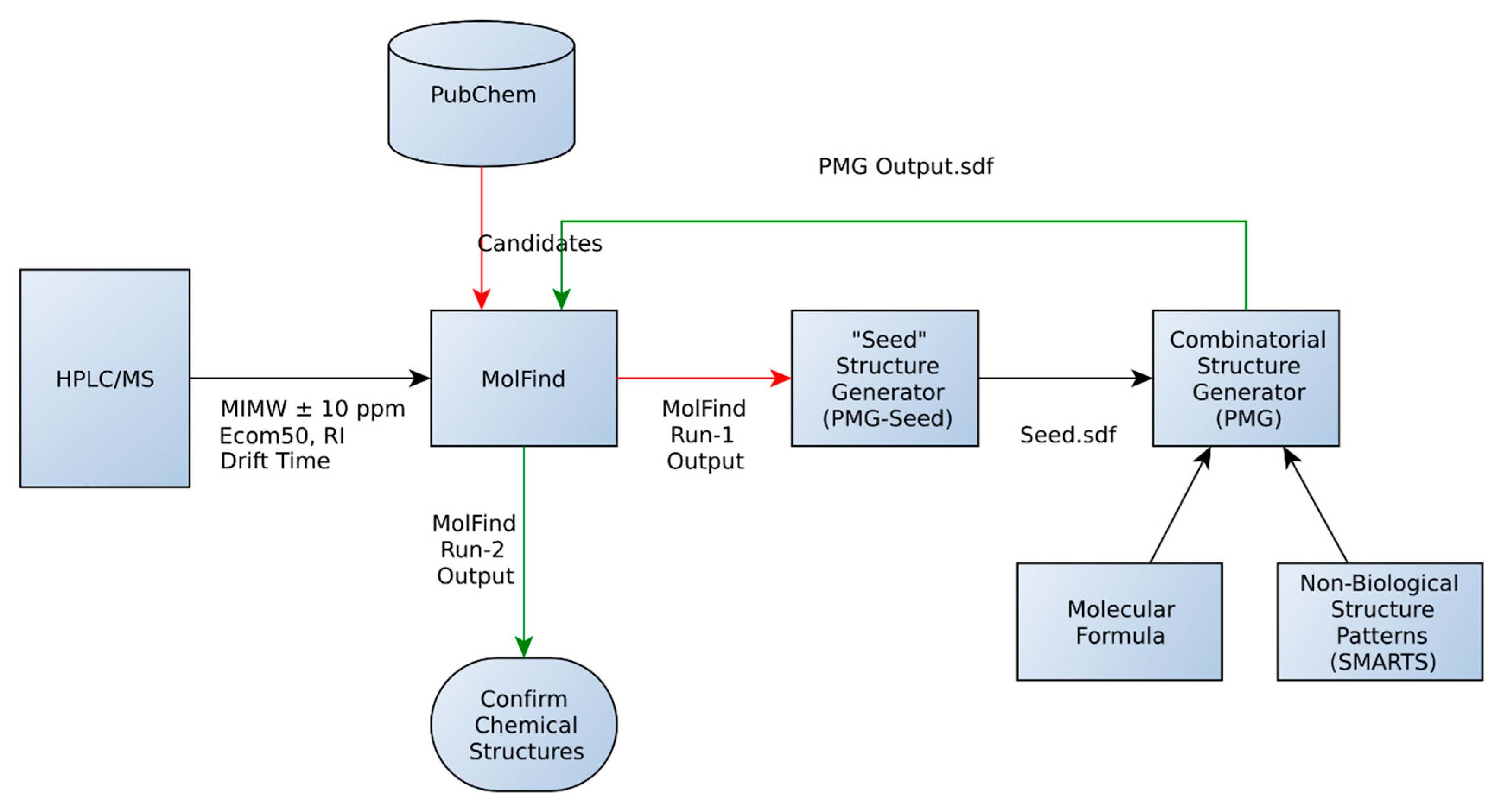
:
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
- (1)
- An initial MolFind [20] run using PubChem. Mono isotopic molecular weight (MIMW), retention index (RI), Ecom50, drift time and collision induced dissociation spectra (CID) of an unknown are inputted into MolFind’s graphical user interface. MolFind’s built-in QSPR filters and CID spectra predictor MetFrag will provide a ranked list of PubChem candidates that best match the input data in JSON and CSV formats. This step ensures that each of the PubChem candidates selected for subsequent processing (step 2) not only has the correct MIMW, but also has an RI, Ecom50, drift time and CID spectrum that closely matches the unknown. Thus, although these compounds are not an exact match to the unknown, they provide a good structural approximation to the unknown. This step also ensures that there are at least some structurally related “hits” in PubChem in order to proceed with seed structure generation.
- (2)
- Output from MolFind (step 1, in JSON format) is fed into the “seed” structure generation program (PMG-Seed); this program will generate one “seed” structure for the combinatorial structure generation program. The seed structure is available in SDF format and represents a consensus structural feature that is shared among the PubChem “hits” identified in step 1 above. For the purposes of this study, during this step the actual unknown and all of its stereoisomers are deleted in order to simulate the unavailability of the unknown in the PubChem database.
- (3)
- Combinatorial structure generation with Parallel Molecular Generator (PMG) [26]; structure generation is controlled by the seed structure and a list of non-endogenous mammalian structures (i.e., structures not allowed in the PMG generated structures). PMG takes the molecular formula, the seed structure (in SDF format) and the list of non-endogenous mammalian structures (in SDF format) as input. PMG’s output is a list of potential unknowns (i.e., candidates) in SDF format all of which have the correct molecular formula, contain the consensus seed structure generated from step 2, and do not contain any non-endogenous mammalian structures.
- (4)
- Finally a second MolFind run is carried out with PMG generated structures from step 3. This step is identical to step 1 above, but instead of filtering PubChem candidates, we are filtering PMG generated candidates using RI, Ecom50, drift time and CID spectra matching. Each of these steps is described in more detail below.
4.1. Initial MolFind Run
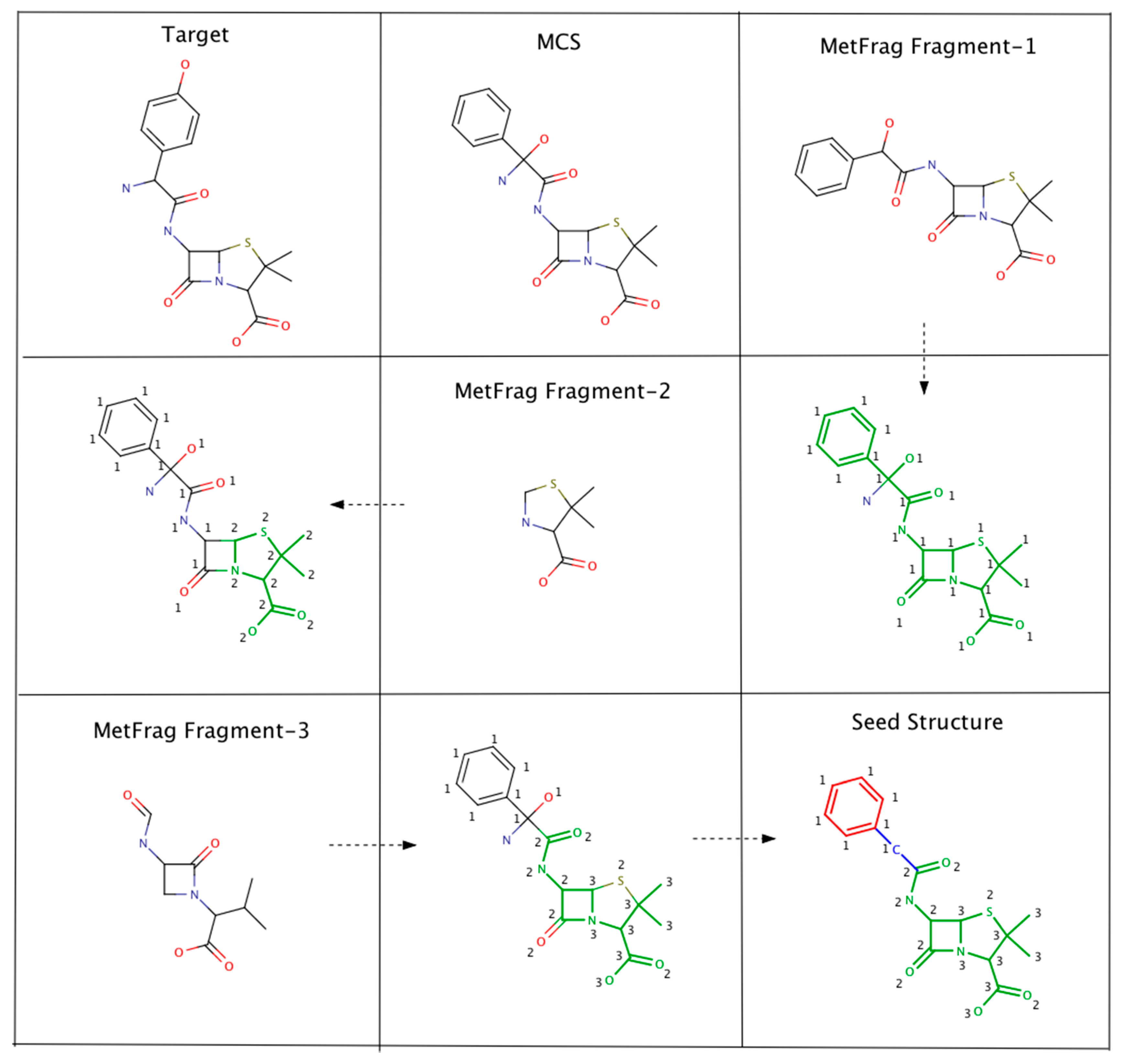
4.2. Seed Structure Generator (PMG-Seed)
- (1)
- Algorithm-1: Top MetFrag Fragment from Filtered Candidates
- (2)
- Algorithm-2: Top MetFrag Fragment from Top PubChem Cluster
- (3)
- Algorithm-3: Intersecting MetFrag Fragments
4.2.1. Algorithm-1: Top MetFrag Fragment from Filtered Candidates
4.2.2. Algorithm-2: Top MetFrag Fragment from Top PubChem Cluster
- RI Window = Maximum RI value deviation based on model statistics [20] (compounds that are within the experimental RI window are considered potential candidates);
- ΔEcom50 Window = Maximum Ecom50 deviation based on model statistics [20] (compounds that are within Experimental Ecom50 ± Ecom50 window are considered potential candidates);
- ΔDrift Time= Experimental Drift Time – Predicted Drift Time;
- Drift Time Window = Maximum Drift Time deviation based on model statistics [20];
4.2.3. Algorithm-3: Intersecting MetFrag Fragments
4.3. Combinatorial Structure Generation
4.4. Refiltering PMG Generated Structures
4.5. Test Dataset and Calculations
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Pon, A.; Wilson, M.; Greiner, R.; Wishart, D. CFM-ID: A web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014, 42, W94–W99. [Google Scholar] [CrossRef] [PubMed]
- Ridder, L.; van der Hooft, J.J.; Verhoeven, S.; de Vos, R.C.; Bino, R.J.; Vervoort, J. Automatic chemical structure annotation of an LC-MSn based metabolic profile from green tea. Anal. Chem. 2013, 85, 6033–6040. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kora, G.; Bowen, B.P.; Pan, C. MIDAS: A database-searching algorithm for metabolite identification in metabolomics. Anal. Chem. 2014, 86, 9496–9503. [Google Scholar] [CrossRef] [PubMed]
- Mak, T.D.; Laiakis, E.C.; Goudarzi, M.; Fornace, A.J., Jr. Selective paired ion contrast analysis: A novel algorithm for analyzing postprocessed LC-MS metabolomics data possessing high experimental noise. Anal. Chem. 2015, 87, 3177–3186. [Google Scholar] [CrossRef] [PubMed]
- Stupp, G.S.; Clendinen, C.S.; Ajredini, R.; Szewc, M.A.; Garrett, T.; Menger, R.F.; Yost, R.A.; Beecher, C.; Edison, A.S. Isotopic ratio outlier analysis global metabolomics of Caenorhabditis elegans. Anal. Chem. 2013, 85, 11858–11865. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Reymond, J.L.; Ruddigkeit, L.; Blum, L.; van Deursen, R. The enumeration of chemical space. Wires Comput. Mol. Sci. 2012, 2, 717–733. [Google Scholar] [CrossRef]
- Peironcely, J.E.; Rojas-Cherto, M.; Fichera, D.; Reijmers, T.; Coulier, L.; Faulon, J.L.; Hankemeier, T. OMG: Open molecule generator. J. Cheminform. 2012, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Gugisch, R.; Kerber, A.; Laue, R.; Meringer, M.; Rucker, C. MOLGEN-CID—A canonizer for molecules and graphs accessible through the Internet. J. Chem. Inform. Comput. Sci. 2004, 44, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Benecke, C.; Grund, R.; Hohberger, R.; Kerber, A.; Laue, R.; Wieland, T. Molgen(+), a Generator of Connectivity Isomers and Stereoisomers for Molecular-Structure Elucidation. Anal. Chim. Acta 1995, 314, 141–147. [Google Scholar] [CrossRef]
- Rojas-Cherto, M.; Peironcely, J.E.; Kasper, P.T.; van der Hooft, J.J.; de Vos, R.C.; Vreeken, R.; Hankemeier, T.; Reijmers, T. Metabolite identification using automated comparison of high-resolution multistage mass spectral trees. Anal. Chem. 2012, 84, 5524–5534. [Google Scholar] [CrossRef] [PubMed]
- Meringer, M.; Schymanski, E.L. Small Molecule Identification with MOLGEN and Mass Spectrometry. Metabolites 2013, 3, 440–462. [Google Scholar] [CrossRef] [PubMed]
- Schymanski, E.L.; Gallampois, C.M.; Krauss, M.; Meringer, M.; Neumann, S.; Schulze, T.; Wolf, S.; Brack, W. Consensus structure elucidation combining GC/EI-MS, structure generation, and calculated properties. Anal. Chem. 2012, 84, 3287–3295. [Google Scholar] [CrossRef] [PubMed]
- Peironcely, J.E.; Rojas-Chertó, M.; Tas, A.; Vreeken, R.J.; Reijmers, T.; Coulier, L.; Hankemeier, T. Automated Pipeline for de novo Metabolite Identification Using Mass Spectrometry-Based Metabolomics. Anal. Chem. 2013, 85, 3576–3583. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Cherto, M.; van Vliet, M.; Peironcely, J.E.; van Doorn, R.; Kooyman, M.; te Beek, T.; van Driel, M.A.; Hankemeier, T.; Reijmers, T. MetiTree: A web application to organize and process high-resolution multi-stage mass spectrometry metabolomics data. Bioinformatics 2012, 28, 2707–2709. [Google Scholar] [CrossRef] [PubMed]
- Menikarachchi, L.C.; Cawley, S.; Hill, D.W.; Hall, L.M.; Hall, L.; Lai, S.; Wilder, J.; Grant, D.F. MolFind: A software package enabling HPLC/MS-based identification of unknown chemical structures. Anal. Chem. 2012, 84, 9388–9394. [Google Scholar] [CrossRef] [PubMed]
- Hamdalla, M.A.; Mandoiu, I.I.; Hill, D.W.; Rajasekaran, S.; Grant, D.F. BioSM: Metabolomics tool for identifying endogenous mammalian biochemical structures in chemical structure space. J. Chem. Inf. Model. 2013, 53, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.; Schmidt, S.; Muller-Hannemann, M.; Neumann, S. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinform. 2010, 11, 148. [Google Scholar] [CrossRef] [PubMed]
- ChemAxon, 5.4.1.1. Available online: http://www.chemaxon.com (accessed on 25 January 2014).
- Hill, D.W.; Kertesz, T.M.; Fontaine, D.; Friedman, R.; Grant, D.F. Mass spectral metabonomics beyond elemental formula: Chemical database querying by matching experimental with computational fragmentation spectra. Anal. Chem. 2008, 80, 5574–5582. [Google Scholar] [CrossRef] [PubMed]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Jaghoori, M.M.; Jongmans, S.-S.T.Q.; de Boer, F.; Peironcely, J.; Faulon, J.-L.; Reijmers, T.; Hankemeier, T. PMG: Multi-core Metabolite Identification. Electron. Notes Theor. Comput. Sci. 2013, 299, 53–60. [Google Scholar] [CrossRef]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. The Chemistry Development Kit (CDK): An open-source Java library for Chemo- and Bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, C.; Hoppe, C.; Kuhn, S.; Floris, M.; Guha, R.; Willighagen, E.L. Recent developments of the chemistry development kit (CDK)—An open-source java library for chemo- and bioinformatics. Curr. Pharm. Des. 2006, 12, 2111–2120. [Google Scholar] [CrossRef] [PubMed]
- Hac - A Java Class Library for Hierarchical Agglomerative Clustering. Available online: http://sape.inf.usi.ch (accessed on 16 May 2013).
- Albaugh, D.R.; Hall, L.M.; Hill, D.W.; Kertesz, T.M.; Parham, M.; Hall, L.H.; Grant, D.F. Prediction of HPLC retention index using artificial neural networks and IGroup E-state indices. J. Chem. Inf. Model. 2009, 49, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Hall, L.M.; Hall, L.H.; Kertesz, T.M.; Hill, D.W.; Sharp, T.R.; Oblak, E.Z.; Dong, Y.W.; Wishart, D.S.; Chen, M.H.; Grant, D.F. Development of Ecom50 and retention index models for nontargeted metabolomics: Identification of 1,3-dicyclohexylurea in human serum by HPLC/mass spectrometry. J. Chem. Inf. Model. 2012, 52, 1222–1237. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.W.; Baveghems, C.L.; Albaugh, D.R.; Kormos, T.M.; Lai, S.; Ng, H.K.; Grant, D.F. Correlation of Ecom50 values between mass spectrometers: Effect of collision cell radiofrequency voltage on calculated survival yield. Rapid Commun. Mass Spectrom. 2012, 26, 2303–2310. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, T.M.; Hall, L.H.; Hill, D.W.; Grant, D.F. CE50: Quantifying collision induced dissociation energy for small molecule characterization and identification. J. Am. Soc. Mass Spectrom. 2009, 20, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Read, R.C. Every one a winner or how to avoid isomorphism search when cataloguing combinatorial configurations. Ann. Discret. Math. 1978, 2, 107–120. [Google Scholar]
- McKay, B.D. Isomorph-free exhaustive generation. J. Algorithms 1998, 26, 306–324. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Algorithm | Number of Correct Seed Structures (/39) | Average % Seed Similarity | % Seed Similarity Range |
|---|---|---|---|
| Algorithm-1 | 24 | 49.5 | 31.8–76.9 |
| Algorithm-2 | 19 | 49.7 | 15.7–87.5 |
| Algorithm-3–1 | 13 | 71.4 | 29.4–90.9 |
| Algorithm-3–2 | 13 | 71.4 | 29.4–90.9 |
| Algorithm-3–3 | 20 | 66.1 | 29.4–92.0 |
| Algorithm-3–4 | 14 | 63.7 | 29.4–90.9 |
| Algorithm-3–5 | 18 | 69.0 | 37.5–92.0 |
| Algorithm-3–6 | 21 | 66.7 | 37.5–89.5 |
| Target | PMG-Seed | Number of PMG Structures | Number after MolFind | MetFrag Score Rank of the Correct Structure |
|---|---|---|---|---|
 |  | 39482 | 146 | 11 |
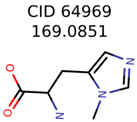 | 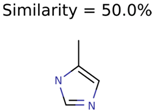 | 58737 | 1502 | 8 |
 |  | 34891 | 2889 | 1230 |
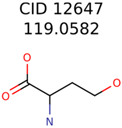 | 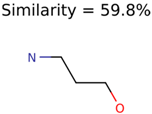 | 644 | 40 | 32 |
 | 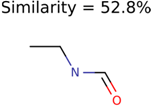 | 230 | 33 | 1 |
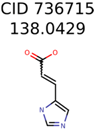 | 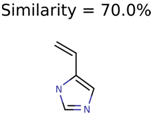 | 289 | 2 | 2 |
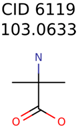 | 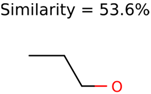 | 409 | 15 | 1 |
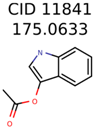 |  | 1726 | 99 | 3 |
 |  | 922 | 30 | 16 |
| Target | PMG-Seed | Number of PMG Structures | Number after MolFind | MetFrag Score Rank of the Correct Structure |
|---|---|---|---|---|
 | 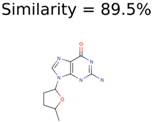 | 38 | 2 | 2 |
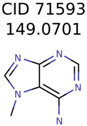 | 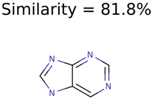 | 20 | 3 | 1 |
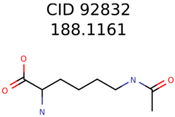 | 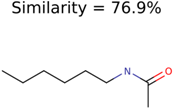 | 7965 | 1638 | 106 |
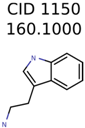 | 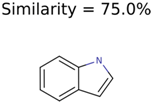 | 266 | 8 | 7 |
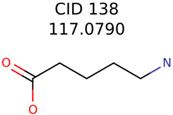 |  | 285 | 48 | 43 |
 |  | 11 | 4 | 2 |
 | 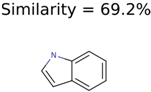 | 7706 | 531 | 24 |
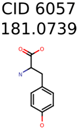 | 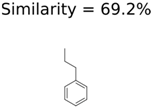 | 13957 | 1124 | 354 |
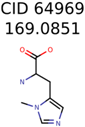 | 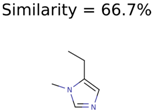 | 8201 | 288 | 5 |
 | 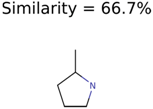 | 437 | 22 | 13 |
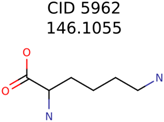 | 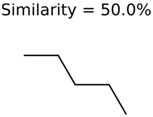 | 25951 | 1001 | 188 |
| Target PubChem ID | Number of Structures | MetFrag Score Ranking of the Correct Structure | ||
|---|---|---|---|---|
| Before MM Filter * | After MM Filter | Before MM Filter * | After MM Filter | |
| 187790 | 2 | 2 | 2 | 2 |
| 71593 | 3 | 3 | 1 | 1 |
| 92832 | 1638 | 367 | 106 | 34 |
| 1150 | 8 | 8 | 7 | 7 |
| 138 | 48 | 26 | 43 | 26 |
| 3134 | 4 | 4 | 2 | 2 |
| 11841 | 531 | 49 | 24 | 16 |
| 6057 | 1124 | 421 | 354 | 148 |
| 64969 | 288 | 163 | 5 | 5 |
| 825 | 22 | 20 | 13 | 13 |
| 5962 | 1001 | 211 | 188 | Filtered Out |
| Variant | MetFrag Fragment Set | Atom Deletion Scheme |
|---|---|---|
| Algorithm-3–1 | Top cluster | Retain MCS atoms with at least 1 match |
| Algorithm-3–2 | All candidates | Retain MCS atoms with at least 1 match |
| Algorithm-3–3 | Top cluster | Retain MCS atoms with at least 2 matches |
| Algorithm-3–4 | All candidates | Retain MCS atoms with at least 2 matches |
| Algorithm-3–5 | Top cluster | Retain MCS atoms with at least average number of atom matches * |
| Algorithm-3–6 | All candidates | Retain MCS atoms with at least average number of atom matches * |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menikarachchi, L.C.; Dubey, R.; Hill, D.W.; Brush, D.N.; Grant, D.F. Development of Database Assisted Structure Identification (DASI) Methods for Nontargeted Metabolomics. Metabolites 2016, 6, 17. https://doi.org/10.3390/metabo6020017
Menikarachchi LC, Dubey R, Hill DW, Brush DN, Grant DF. Development of Database Assisted Structure Identification (DASI) Methods for Nontargeted Metabolomics. Metabolites. 2016; 6(2):17. https://doi.org/10.3390/metabo6020017
Chicago/Turabian StyleMenikarachchi, Lochana C., Ritvik Dubey, Dennis W. Hill, Daniel N. Brush, and David F. Grant. 2016. "Development of Database Assisted Structure Identification (DASI) Methods for Nontargeted Metabolomics" Metabolites 6, no. 2: 17. https://doi.org/10.3390/metabo6020017
APA StyleMenikarachchi, L. C., Dubey, R., Hill, D. W., Brush, D. N., & Grant, D. F. (2016). Development of Database Assisted Structure Identification (DASI) Methods for Nontargeted Metabolomics. Metabolites, 6(2), 17. https://doi.org/10.3390/metabo6020017