Clinically Important Features of Porphyrin and Heme Metabolism and the Porphyrias

Abstract
:1. Introduction
2. Key Features of Heme Metabolism and Regulation
2.1. Heme Biosynthesis and Its Regulation
2.1.1. Heme Biosynthesis
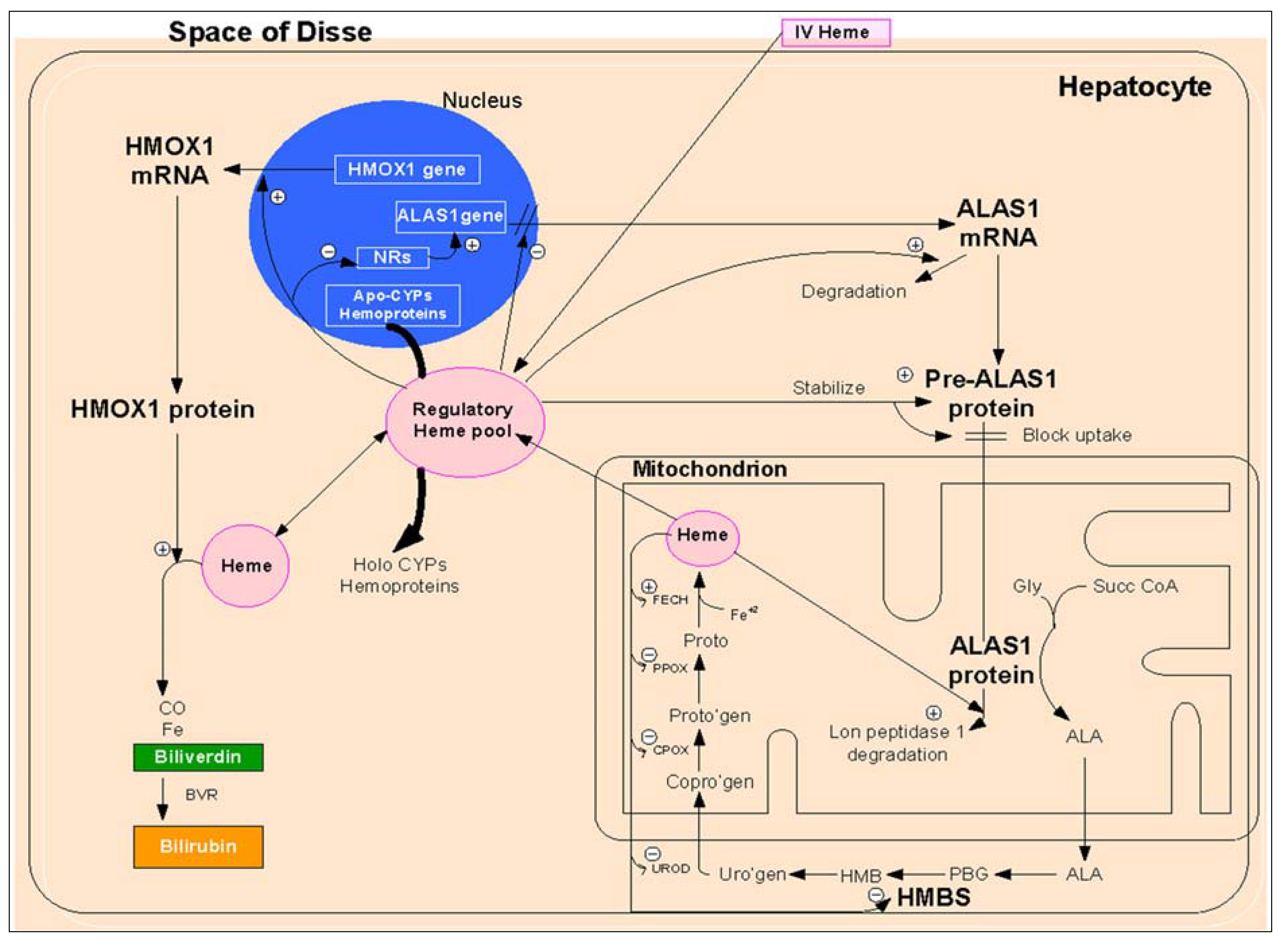
2.1.1.1. Regulation of Heme Biosynthesis by ALA Synthase

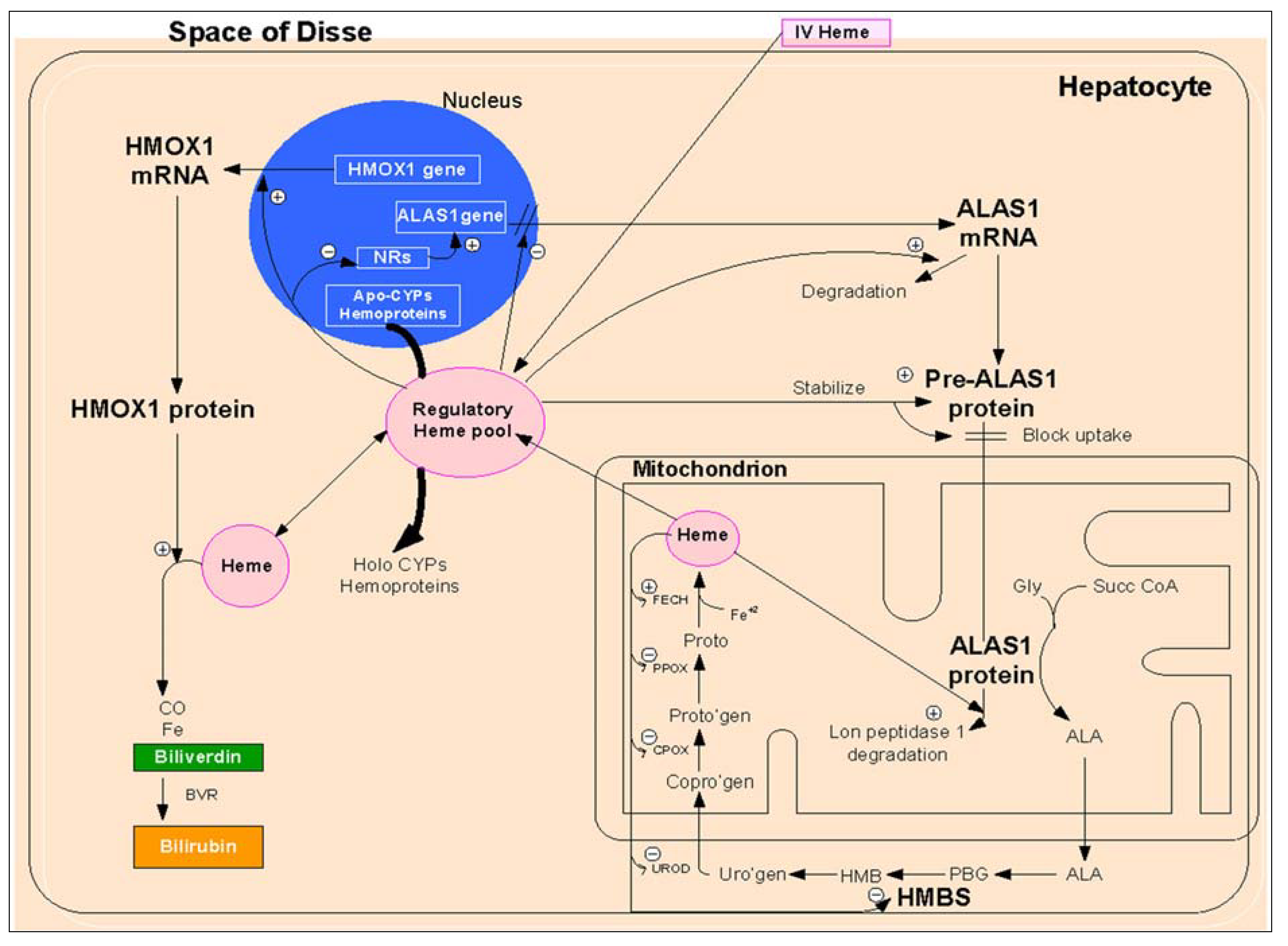{kind=link}
| Rate-Controlling Enzyme | Tissue Origin Subcellular Location | Gene and Chromosome Location | Gene Regulation |
|---|---|---|---|
| Heme Biosynthesis ALAS1 ALAS2 | Ubiquitous Mitochondria Bone marrow Mitochondria | ALAS1 3p31.2 ALAS2 Xp11.2 | Transcriptional regulation: Down-regulation by heme, glucose and sugars Induction by chemical, drugs, stress, circadian rhythm Post-transcriptional regulation: Destabilization of ALAS1 mRNA by heme Post-translational regulation: Impediment to pre-ALAS1 import into mitochondria Degradation of ALAS1 protein by heme Up-regulation by hypoxia and iron |
| Heme Catabolism HMOX1 HMOX2 | Ubiquitous Mainly in smooth endoplasmic reticulum, mitochondria and nucleus Brain and testes Smooth endoplasmic reticulum | HMOX1 22q12.3 | Transcriptional regulation: Up-regulation by heme and other metalloporphyrins Down-regulation by metalloporphyrins Induction by chemical and physical stresses Genetic polymorphisms Translational regulation: miRNAs Alternative splicing in the 5′-UTR |
2.1.1.2. Transcriptional Regulation
2.1.1.3. Post-Transcriptional Regulation
2.1.1.4. Post-Translational Regulation
2.2. Heme Catabolism and Its Regulation
2.2.1. Heme Catabolism
2.2.2. Regulation of HMOX1
2.2.2.1. Transcriptional Regulation
2.2.2.2. Translational Regulation
3. The Porphyrias: Disorders of Heme Synthesis
3.1. Acute Porphyrias: Acute Intermittent Porphyria as a Paradigm
3.1.1. Epidemiology
| A. According to the Clinical Manifestations of Disease | |||||
| Type | Inheritance | Gene Affected | Chromosomal Location | Comments | |
| Acute or Inducible Porphyrias | ALADP | AR | Pbgs | 9q34 | Very rare severe disease in infancy |
| AIP | AD | Hmbs | 11q23.3 | Most severe form | |
| HCP | AD | Cpox | 3q12 | May also have cutaneous features | |
| VP | AD | Ppox | 1q22 | May also have cutaneous features | |
| Cutaneous Chronic Porphyrias | CEP | AR | Uro3 | 10q26.1-q26.2 | Rare usually manifests itself in infancy/childhood |
| HEP | AR | Urod | 1p34.1 | Rare usually manifests itself in infancy/childhood | |
| PCT (Type I) | Acquired | None | Diseases of adults | ||
| PCT (Type II) | AD | Urod | 1p34.1 | Requires additional defects | |
| EPP | AR | Fech | 18q21.31 | Common; onset in infancy | |
| XLPP | X-linked | Alas1 | Xp11.21 | Gain of function mutations | |
| B. According to the Clinical Manifestations of Disease | |||||
| Type | Inheritance | Gene Affected | Chromosomal Location | Comments | |
| Acute Hepatic Porphyrias | ALADP | AR | Pbgs | 9q34 | Very rare severe disease in infancy |
| AIP | AD | Hmbs | 11q23.3 | Most severe form | |
| HCP | AD | Cpox | 3q12 | May also have cutaneous features | |
| VP | AD | Ppox | 1q22 | May also have cutaneous features | |
| Chronic Hepatic Porphyrias | PCT (Type I) | Acquired | None | Diseases of adults | |
| PCT (Type II) | AD | Urod | 1p34.1 | Requires additional defects | |
| HEP | AR | Urod | 1p34.1 | Rare usually manifests itself in infancy/childhood | |
| Erythropoietic Porphyrias | CEP | AR | Uro3 | 10q26.1-q26.2 | Rare usually manifests itself in infancy/childhood |
| EPP | AR | Fech | 18q21.31 | Common; onset in infancy | |
3.1.2. Pathogenesis of Acute Attacks
| Exacerbating Factors | Common Unsafe Drugs |
|---|---|
| Drugs and chemicals—especially | ➢ Excess alcohol |
| ➢ Barbiturates | |
| ➢ Estrogens | |
| ➢ Hydantoins | |
| ➢ Progestagens | |
| ➢ Sulfonamides | |
| ➢ All drugs that are suicide substrates or potent inducers of cytochrome P450 | |
| Dieting; fasting; deficiency of carbohydrate intake (gastric bypass surgery) | - |
| Exhaustion—emotional or physical | - |
| Intercurrent acute illnesses | - |
3.1.3. Clinical Features
3.2. Diagnosis
| Symptoms and Signs | Estimated Symptoms and Signs Incidence, (%) | Comment | |
|---|---|---|---|
| Gastrointestinal | |||
| Abdominal Pain | 85–95 | Usually unremitting (for hours or longer) and poorly localized, but can be cramping. | |
| Vomiting | 43–88 | Neurologic in origin and rarely accompanied by peritoneal signs, fever or leukocytosis. Nausea vomiting often accompanies abdominal pain. | |
| Constipation | 48–84 | May be accompanied by bladder paresis. | |
| Neurologic | |||
| Pain in extremities and/or back | 50–70 | Pain may begin in the chest or back and move to the abdomen. Extremity pain, chest, neck or back indicates involvement of sensory nerves, with objective sensory loss reported in 10%–40% of cases. | |
| Paresis | 42–68 | May occur early or late during a severe attack. | |
| Respiratory Paralysis | 9–20 | Muscle weakness usually begins proximally rather than distally and more often in the upper than lower extremities, preceded by progressive peripheral motor neuropathy and paresis. | |
| Mental Symptoms | 40–58 | May range from minor behavioral changes to agitation, confusion, hallucinations and depression | |
| Convulsions | 10–20 | A central neurologic manifestation of porphyria or due to hyponatremia, which often results from syndrome of inappropriate antidiuretic hormone secretion or sodium depletion. | |
| Cardiovascular | |||
| Tachycardia | 64–85 | May warrant treatment to control rate, if symptomatic. | |
| Systemic arterial hypertension | 36–55 | May require treatment during acute attacks and may sometimes become chronic. | |
Management
| Type of Porphyria | Enzyme Defect | Urine | Stool | Plasma | RBCs |
|---|---|---|---|---|---|
| X-linked protoporphyria | ALA synthase-2 (gain of function) | Normal | PROTO | PROTO | Zn PROTO |
| ALA dehydratase deficiency (ADP) | ALA dehydratase | COPRO ALA | Normal | ALA | Zn PROTO |
| Acute intermittent porphyria (AIP) | PBG deaminase | ALA, PBG, URO I | Normal COPRO I | ALA, PBG, URO I | ↓PBGD |
| Congenital erythropoietic porphyria (CEP) | Uroporphyrinogen III synthase (cosynthase) | COPRO I URO I | COPRO I | COPRO I URO I | COPRO I URO I |
| Porphyria cutanea tarda (PCT) and Hepatoerythropoietic porphyria (HEP) | Uroporphyrinogen III decarboxylase | Uroporphyrin, heptacarboxyl porphyrin | Heptacarboxyl porphyrin ISOCOPRO | Uroporphyrin, heptacarboxyl porphyrin | Zn PROTO |
| Hereditary coproporphyria (HCP) | Coproporphyrinogen III oxidase | ALA, PBG, COPRO III | COPRO III | COPRO | Normal |
| Variegate porphyria (VP) | Protoporphyrinogen oxidase | ALA, PBG, COPRO III | PROTO COPRO III | Porphyrin peptide conjugate | Normal |
| Erythropoietic protoporphyria (EPP) | Ferrochelatase | COPRO III with hepatopathy | PROTO | PROTO | PROTO |
3.3. Hereditary Coproporphyria (HCP)
3.3.1. Pathogenesis
3.3.2. Clinical Features
3.3.3. Diagnosis
3.3.4. Management
3.4. Variegate Porphyria (VP)
3.4.1. Pathogenesis
3.4.2. Clinical Features
3.4.3. Diagnosis
3.4.4. Management
3.5. 5-Aminolevulinic Acid Dehydratase Deficient Porphyria (ALADP)
3.5.1. Pathogenesis
3.5.2. Clinical Features
3.5.3. Diagnosis
3.5.4. Management
3.6. Chronic Hepatic Porphyrias
3.6.1. Porphyria Cutanea Tarda (PCT)
3.6.1.1. Epidemiology
3.6.1.2. Classification of PCT
3.6.1.3. Pathogenesis
3.6.1.4. Clinical Features
| Exacerbating Factors | Common Drugs/Chemical Triggers |
|---|---|
| Alcohol excess with alcoholic liver disease | |
| Chronic hepatitis C | |
| Human immunodeficiency virus infection | |
| Mutations in the Hfe, Hjv and Tfr2 | |
| End stage renal disease | |
| Drugs and chemicals, but especially | ➢ Excess alcohol |
| ➢ Estrogens | |
| ➢ Polyhalogenated aromatic chemicals |
3.6.1.5. Diagnosis
3.6.1.6. Management
3.7. The Erythropoietic Porphyrias
3.7.1. Congenital Erythropoietic Porphyria (CEP)
3.7.1.1. Pathogenesis
3.7.1.2. Clinical Features (Table 5)
3.7.1.3. Diagnosis
3.7.1.4. Management
3.7.2. Erythropoietic Protoporphyria (EPP, XLPP)
3.7.2.1. Pathogenesis
3.7.2.2. Clinical Features
3.7.2.3. Diagnosis (Table 5)
3.7.2.4. Management
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Li, M.; Vizzard, M.A.; Jaworski, D.M.; Galbraith, R.A. The weight loss elicited by cobalt protoporphyrin is related to decreased activity of nitric oxide synthase in the hypothalamus. J. Appl. Physiol. 2006, 100, 1983–1991. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, R.A.; Hodgdon, I.; Grimm, M.S.; Vizzard, M.A. Prolonged retention of the anorectic cobalt protoporphyrin in the hypothalamus and the resulting expression of Fos. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R465–R471. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Shan, Y.; Lambrecht, R.W.; Donohue, S.E.; Bonkovsky, H.L. Differential regulation of human ALAS1 mRNA and protein levels by heme and cobalt protoporphyrin. Mol. Cell Biochem. 2008, 319, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Kolluri, S.; Sadlon, T.J.; May, B.K.; Bonkovsky, H.L. Haem repression of the housekeeping 5-aminolaevulinic acid synthase gene in the hepatoma cell line LMH. Biochem. J. 2005, 392, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Lin, J.; Rhee, J.; Peyer, A.K.; Chin, S.; Wu, P.H.; Meyer, U.A.; Spiegelman, B.M. Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1alpha. Cell 2005, 122, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [PubMed]
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.; Verspohl, F. The “glucose effect” in acute hepatic porphyrias and in experimental porphyria. Klin. Wochenschr. 1981, 59, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Scassa, M.E.; Guberman, A.S.; Ceruti, J.M.; Cánepa, E.T. Hepatic nuclear factor 3 and nuclear factor 1 regulate 5-aminolevulinate synthase gene expression and are involved in insulin repression. J. Biol. Chem. 2004, 279, 28082–28092. [Google Scholar] [CrossRef] [PubMed]
- Scassa, M.E.; Guberman, A.S.; Varone, C.L.; Cánepa, E.T. Phosphatidylinositol 3-kinase and Ras/mitogen-activated protein kinase signaling pathways are required for the regulation of 5-aminolevulinate synthase gene expression by insulin. Exp. Cell Res. 2001, 271, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.E.; Bloomer, J.R.; Bonkovsky, H.L.; Kushner, J.P.; Pierach, C.A.; Pimstone, N.R.; Desnick, R.J. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann. Intern. Med. 2005, 142, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.D.; Kushner, J.P. Fast track to the porphyrias. Nat. Med. 2005, 11, 1049–1050. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Yin, L.; Hanniman, E.A.; Joshi, S.; Lazar, M.A. Negative feedback maintenance of heme homeostasis by its receptor, Rev-erbalpha. Genes Dev. 2009, 23, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.J.; Podvinec, M.; Kaufmann, M.R.; Meyer, U.A. Drugs mediate the transcriptional activation of the 5-aminolevulinic acid synthase (ALAS1) gene via the chicken xenobiotic-sensing nuclear receptor (CXR). J. Biol. Chem. 2002, 277, 34717–34726. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.J.; Zumsteg, A.; Meyer, U.A. Nuclear receptors constitutive androstane receptor and pregnane X receptor activate a drug-responsive enhancer of the murine 5-aminolevulinic acid synthase gene. J. Biol. Chem. 2003, 278, 39392–39401. [Google Scholar] [CrossRef] [PubMed]
- Podvinec, M.; Handschin, C.; Looser, R.; Meyer, U.A. Identification of the xenosensors regulating human 5-aminolevulinate synthase. Proc. Natl. Acad. Sci. USA 2004, 101, 9127–9132. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.G.; Elder, G.H. Alternative splicing and tissue-specific transcription of human and rodent ubiquitous 5-aminolevulinate synthase (ALAS1) genes. Biochim. Biophys. Acta 2001, 1518, 95–105. [Google Scholar]
- Roberts, A.G.; Redding, S.J.; Llewellyn, D.H. An alternatively-spliced exon in the 5′-UTR of human ALAS1 mRNA inhibits translation and renders it resistant to haem-mediated decay. FEBS Lett. 2005, 579, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Lathrop, J.T.; Timko, M.P. Regulation by heme of mitochondrial protein transport through a conserved amino acid motif. Science 1993, 259, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Dailey, T.A.; Woodruff, J.H.; Dailey, H.A. Examination of mitochondrial protein targeting of haem synthetic enzymes: In vivo identification of three functional haem-responsive motifs in 5-aminolaevulinate synthase. Biochem. J. 2005, 386, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Munakata, H.; Munakata, H.; Sun, J.Y.; Yoshida, K.; Nakatani, T.; Honda, E.; Hayakawa, S.; Furuyama, K.; Hayashi, N. Role of the heme regulatory motif in the heme-mediated inhibition of mitochondrial import of 5-aminolevulinate synthase. J. Biochem. 2004, 136, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Li, T.; Hou, W.; Zheng, J.; Schrum, L.W.; Bonkovsky, H.L. Lon peptidase 1 (LONP1)-dependent breakdown of mitochondrial 5-aminolevulinic acid synthase protein by heme in human liver cells. J. Biol. Chem. 2011, 286, 26424–26430. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Sun, J.; Taketani, S.; Nakajima, O.; Nishitani, C.; Sassa, S.; Hayashi, N.; Yamamoto, M.; Shibahara, S.; Fujita, H.; et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 2001, 20, 2835–2843. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Lambrecht, R.W.; Ghaziani, T.; Donohue, S.E.; Bonkovsky, H.L. Role of Bach-1 in regulation of heme oxygenase-1 in human liver cells: Insights from studies with small interfering RNAS. J. Biol. Chem. 2004, 279, 51769–51774. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Shan, Y.; Zheng, J.; Lambrecht, R.W.; Donohue, S.E.; Bonkovsky, H.L. Zinc mesoporphyrin induces rapid and marked degradation of the transcription factor Bach1 and up-regulates HO-1. Biochim. Biophys. Acta 2008, 1779, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Camhi, S.; Choi, A. Identification of a second region upstream of the mouse heme oxygenase-1 gene that functions as a basal level and inducer-dependent transcription enhancer. J. Biol. Chem. 1995, 270, 11977–11984. [Google Scholar] [CrossRef] [PubMed]
- Camhi, S.L.; Alam, J.; Otterbein, L.; Sylvester, S.L.; Choi, A.M. Induction of heme oxygenase-1 gene expression by lipopolysaccharide is mediated by AP-1 activation. Am. J. Respir. Cell Mol. Biol. 1995, 13, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Tian, Q.; Zheng, J.; Bonkovsky, H.L. MicroRNA-196 represses Bach1 protein and hepatitis C virus gene expression in human hepatoma cells expressing hepatitis C viral proteins. Hepatology 2010, 51, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Mukherjee, N.; Sachse, C.; Frenzel, C.; Majoros, W.H.; Chi, J.T.; Braich, R.; Manoharan, M.; Soutschek, J.; Ohler, U.; et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature 2007, 450, 1096–1099. [Google Scholar] [CrossRef] [PubMed]
- Pulkkinen, K.H.; Yla-Herttuala, S.; Levonen, A.L. Heme oxygenase 1 is induced by miR-155 via reduced BACH1 translation in endothelial cells. Free Radic. Biol. Med. 2011, 51, 2124–2131. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Tian, Q.; Steuerwald, N.M.; Schrum, L.W.; Bonkovsky, H.L. The let-7 microRNA enhances heme oxygenase-1 by suppressing Bach1 and attenuates oxidant injury in human hepatocytes. Biochim. Biophys. Acta 2012, 1819, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Lin, S.J.; Lin, M.W.; Tsai, H.L.; Kuo, S.S.; Chen, J.W.; Charng, M.J.; Wu, T.C.; Chen, L.C.; Ding, Y.A.; et al. Microsatellite polymorphism in promoter of heme oxygenase-1 gene is associated with susceptibility to coronary artery disease in type 2 diabetic patients. Hum. Genet. 2002, 111, 1–8. [Google Scholar]
- Kaneda, H.; Ohno, M.; Taguchi, J.; Togo, M.; Hashimoto, H.; Ogasawara, K.; Aizawa, T.; Ishizaka, N.; Nagai, R. Heme oxygenase-1 gene promoter polymorphism is associated with coronary artery disease in Japanese patients with coronary risk factors. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Exner, M.; Minar, E.; Wagner, O.; Schillinger, M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radic. Biol. Med. 2004, 37, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.W.; Sheu, W.H.; Lee, W.L.; Lee, I.T.; Lin, S.Y.; Ting, C.T.; Lee WJ. Shorter, G.T. Repeats in the heme oxygenase-1 gene promoter are associated with a lower severity score in coronary artery disease. J. Chin. Med. Assoc. 2013, 76, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Yamaya, M.; Okinaga, S.; Nakayama, K.; Sekizawa, K.; Shibahara, S.; Sasaki, H. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to emphysema. Am. J. Hum. Genet. 2000, 66, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Bonkovsky, H.L.; Lambrecht, R.W.; Naishadham, D. Genetic variations in heme oxygenase-1 and chronic hepatitis. Hepatology 2010, 52, 400–401. [Google Scholar] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.D.; Chen, C.; Nguyen, J.; Thayanithy, V.; Subramanian, S.; Steer, C.J.; Vercellotti, G.M. Regulation of heme oxygenase-1 protein expression by miR-377 in combination with miR-217. J. Biol. Chem. 2011, 286, 3194–3202. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.; Sponholz, C.; Slaba, M.; Wissuwa, B.; Claus, R.A.; Menzel, U.; Huse, K.; Platzer, M.; Bauer, M. Alternative 5′ untranslated regions are involved in expression regulation of human heme oxygenase-1. PLoS One 2013, 8. Article e77224. [Google Scholar]
- Schmid, R.; Schwartz, S.; Watson, C.J. Porphyrin content of bone marrow and liver in the various forms of porphyria. AMA Arch. Intern. Med. 1954, 93, 167–190. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.; von Tiepermann, R.; Schneider, J.; Schmid, H. New type of hepatic porphyria with porphobilinogen synthase defect and intermittent acute clinical manifestation. Klin. Wochenschr. 1979, 57, 1123–1127. [Google Scholar] [CrossRef]
- Waldenstrom, J. The porphyrias as inborn errors of metabolism. Am. J. Med. 1957, 22, 758–773. [Google Scholar] [CrossRef] [PubMed]
- Tschudy, D.P.; Valsamis, M.; Magnussen, C.R. Acute intermittent porphyria: Clinical and selected research aspects. Ann. Intern. Med. 1975, 83, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Floderus, Y.; Shoolingin-Jordan, P.M.; Harper, P. Acute intermittent porphyria in Sweden. Molecular, functional and clinical consequences of some new mutations found in the porphobilinogen deaminase gene. Clin. Genet. 2002, 62, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.R.; McColl, K.E.L.; Rimington, C.; Goldberg, A. Disorders of Porphyrin Metabolism; Plenum Medical Book Company: New York, NY, USA, 1987. [Google Scholar]
- Mustajoki, P.; Koskelo, P. Hereditary hepatic porphyria in Finland. Acta Med. Scand. 1976, 200, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Yin, X.; Harms, J.; Minder, E.I. Porphyria in Switzerland:15 Years’ experience. Swiss Med. Wkly. 2009, 139, 198–206. [Google Scholar] [PubMed]
- Lamon, J.M.; Frykholm, B.C.; Tschudy, D.P. Family evaluations in acute intermittent porphyria using red cell uropophyrinogen I synthase. J. Med. Genet. 1979, 16, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Von und zu Fraunberg, M.; Pischik, E.; Udd, L.; Kauppinen, R. Clinical and biochemical characteristics and genotype-phenotype correlation in 143 Finnish and Russian patients with acute intermittent porphyria. Medicine 2005, 84, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, Y.; Puy, H.; da Siva, V.; Simonin, S.; Robreau, A.M.; Bonaiti, C.; Phung, L.N.; Deybach, J.-C. Acute intermittent porphyria: The prevalence of mutations in the porphobilinogen deaminase gene in blood donors in France. J. Int. Med. 1997, 242, 213–217. [Google Scholar] [CrossRef]
- Elder, G.; Harper, P.; Badminton, M.; Sandberg, S.; Deybach, J.C. The incidence of inherited porphyrias in Europe. J. Inherit. Metab. Dis. 2013, 36, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Yin, X.; Harms, J.; Minder, E.I. Porphyria in Switzerland, 15 years experience. Swiss Med. Wkly. 2009, 139, 198–206. [Google Scholar] [PubMed]
- Hultdin, J.; Schmauch, A.; Wikberg, A.; Dahlquist, G.; Andersson, C. Acute intermittent porphyria in childhood: A population-based study. Acta Paediatr. 2003, 92, 562–568. [Google Scholar] [CrossRef] [PubMed]
- American Porphyria Foundation. Available online: http://www.porphyriafoundation.com/drug-database (accessed on 29 October 2014).
- European Porphyria Network. Available online: http://www.porphyria-europe.org (accessed on 29 October 2014).
- University of Cape Town Porphyria Service. Available online: http://www.porphyria.uct.ac.za (accessed on 29 October 2014).
- Bonkovsky, H.L.; Guo, J.T.; Hou, W.; Li, T.; Narang, T.; Thapar, M. Porphyrin and heme metabolism and the porphyrias. Compr. Physiol. 2013, 3, 365–401. [Google Scholar] [PubMed]
- Puy, H.; Gouya, L.; Deybach, J.C. Porphyrias. Lancet 2010, 375, 924–937. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, R.; Mustajoki, P. Prognosis of acute porphyria: Occurrence of acute attacks, precipitating factors, and associated diseases. Medicine (Baltim.) 1992, 71, 1–13. [Google Scholar]
- Bonkovsky, H.L.; Maddukuri, V.C.; Yazici, C.; Anderson, K.E.; Bissell, D.M.; Bloomer, J.R.; Phillips, J.D.; Naik, H.; Peter, I.; Baillargeon, G.; et al. Acute Porphyrias in the USA: Features of 108 Subjects from Porphyria Consortium. Am. J. Med. 2014, 9. [Google Scholar] [CrossRef]
- Goldberg, A. Acute intermittent porphyria: A study of 50 cases. Q. J. Med. 1959, 28, 183–209. [Google Scholar] [PubMed]
- Nordmann, Y.; Puy, H. Human hereditary hepatic porphyrias. Clin. Chim. Acta 2002, 325, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Bloomer, J.R.; Bonkovsky, H.L. The porphyrias. Dis Mon. 1989, 35, 1–54. [Google Scholar] [CrossRef] [PubMed]
- Sardh, E.; Andersson, D.E.; Henrichson, A.; Harper, P. Porphyrin precursors and porphyrins in three patients with acute intermittent porphyria and end-stage renal disease under different therapy regimes. Cell. Mol. Biol. 2009, 55, 66–71. [Google Scholar] [PubMed]
- Deacon, A.C.; Peters, T.J. Identification of acute porphyria: Evaluation of a commercial screening test for urinary porphobilinogen. Ann. Clin. Biochem. 1998, 35, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Bonkovsky, H.L. Porphyrin and Heme Metabolism and Porphyrias. In HEPATOLOGY: A Textbook of LIVER DISEASE, 2nd ed.; Zakim, D., Boyer, T.D., Eds.; Saunders: Philadelphia, PA, USA, 1990; pp. 378–424. [Google Scholar]
- Bonkovsky, H.L.; Barnard, G.F. Diagnosis of porphyric syndromes: A practical approach in the era of molecular biology. Semin. Liver Dis. 1998, 18, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.; Bonkovsky, H.L. Disorders of porphyrin metabolism. Diseases of the Liver and Bile Ducts: A Practical Guide to Diagnosis and Treatment, 1 st ed.; Wu, G., Israel, J., Totowa, N.J., Eds.; Humana Press: New York, NY, USA, 1998; pp. 249–272. [Google Scholar]
- The Human Gene Mutation Datbase at the Institute of Medical Genetics in Cardiff. Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 29 October 2014).
- Anderson, K.E.; Bonkovsky, H.L.; Bloomer, J.R.; Shedlofsky, S.I. Reconstitution of hematin for intravenous infusion. Ann. Intern. Med. 2006, 144, 537–538. [Google Scholar] [CrossRef] [PubMed]
- Bonkovsky, H.L.; Healey, J.F.; Lourie, A.N.; Gerron, G.G. Intravenous heme-albumin in acute intermittent porphyria: Evidence for repletion of hepatic hemoproteins and regulatory heme pools. Am. J. Gastroenterol. 1991, 86, 1050–1056. [Google Scholar] [PubMed]
- Dar, F.S.; Asai, K.; Haque, A.R.; Cherian, T.; Rela, M.; Heaton, N. Liver transplantation for acute intermittent porphyria: A viable treatment? Hepatobiliary Pancreat. Dis. Int. 2010, 9, 93–96. [Google Scholar]
- Seth, A.K.; Badminton, M.N.; Mirza, D.; Russell, S.; Elias, E. Liver transplantation for porphyria: Who, when, and how? Liver Transpl. 2007, 13, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Wahlin, S.; Harper, P.; Sardh, E.; Andersson, C.; Andersson, D.E.; Ericzon, B.G. Combined liver and kidney transplantation in acute intermittent porphyria. Transpl. Int. 2010, 23, e18–e21. [Google Scholar] [CrossRef] [PubMed]
- Mercelis, R.; Hassoun, A.; Verstraeten, L.; De Bock, R.; Martin, J.J. Porphyric neuropathy and hereditary delta-aminolevulinic acid dehydratase deficiency in an adult. J. Neurol. Sci. 1990, 95, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Elder, G.H. Porphyria cutanea tarda. Semin. Liver Dis. 1998, 18, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Brady, J.J.; Jackson, H.A.; Roberts, A.G.; Morgan, R.R.; Whatley, S.D.; Rowlands, G.L.; Darby, C.; Shudell, E.; Watson, R.; Paiker, J.; et al. Co-inheritance of mutations in the uroporphyrinogen decarboxylase and hemochromatosis genes accelerates the onset of porphyria cutanea tarda. J. investig. Dermatol. 2000, 115, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Bonkovsky, H.L. Mechanism of iron potentiation of hepatic uroporphyria: Studies in cultured chick embryo liver cells. Hepatology 1989, 10, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.D.; Bergonia, H.A.; Reilly, C.A.; Franklin, M.R.; Kushner, J.P. A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda. Proc. Natl. Acad. Sci. USA 2007, 104, 5079–5084. [Google Scholar] [CrossRef]
- Badminton, M.N.; Elder, G.H. Molecular mechanisms of dominant expression in porphyria. J. Inherit. Metab. Dis. 2005, 28, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Bonkovsky, H.L.; Poh-Fitzpatrick, M.; Pimstone, N.; Obando, J.; Di Bisceglie, A.; Tattrie, C.; Tortorelli, K.; LeClair, P.; Mercurio, M.G.; Lambrecht, R.W. Porphyria cutanea tarda, hepatitis C, and HFE gene mutations in North America. Hepatology 1998, 27, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Gisbert, J.P.; Garcia-Buey, L.; Pajares, J.M.; Moreno-Otero, R. Prevalence of hepatitis C virus infection in porphyria cutanea tarda: Systematic review and meta-analysis. J. Hepatol. 2003, 39, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Fargion, S.; Piperno, A.; Cappellini, M.D.; Sampietro, M.; Fracanzani, A.L.; Romano, R.; Caldarelli, R.; Marcelli, R.; Vecchi, L.; Fiorelli, G. Hepatitis C virus and porphyria cutanea tarda: Evidence of a strong association. Hepatology 1992, 16, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Pätzold, K.; Hahn, L.; Lohmann, J.; Porst, H. Liver cirrhosis and chronic hepatic porphyria. Z Gastroenterol. 1993, 31 (Suppl. S2), 114–115. [Google Scholar] [PubMed]
- Gisbert, J.P.; García-Buey, L.; Alonso, A.; Rubio, S.; Hernández, A.; Pajares, J.M.; García-Díez, A.; Moreno-Otero, R. Hepatocellular carcinoma risk in patients with porphyria cutanea tarda. Eur. J. Gastroenterol. Hepatol. 2004, 16, 698–692. [Google Scholar]
- Ippen, H. Treatment of porphyria cutanea tarda by phlebotomy. Semin. Hematol. 1977, 14, 253–259. [Google Scholar] [PubMed]
- Tavill, AS. Diagnosis and management of hemochromatosis. Hepatology 2001, 33, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.K.; Kormos-Hallberg, C.; Lee, C.; Sadagoparamanujam, V.M.; Grady, J.J.; Freeman, D.H., Jr.; Anderson, K.E. Low-dose hydroxychloroquine is as effective as phlebotomy in treatment of patients with porphyria cutanea tarda. Clin. Gastroenterol. Hepatol. 2012, 10, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, L.; Di Padova, C.; Cainelli, T.; Reseghetti, A.; Di Padova, F.; Rovagnati, P.; Cantoni, L. A comparative trial of desferrioxamine and hydroxychloroquine for treatment of porphyria cutanea tarda in alcoholic patients. Photodermatol 1984, 1, 286–292. [Google Scholar] [PubMed]
- Pandya, A.G.; Nezafati, K.A.; Ashe-Randolph, M.; Yalamanchili, R. Deferasirox for porphyria cutanea tarda: A pilot study. Arch. Dermatol. 2012, 148, 898–901. [Google Scholar] [PubMed]
- Kordac, V.; Semrádová, M. Treatment of porphyria cutanea tarda with chloroquine. Br. J. Dermatol. 1974, 90, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Freesemann, A.; Frank, M.; Sieg, I.; Doss, M.O. Treatment of porphyria cutanea tarda by the effect of chloroquine on the liver. Skin Pharmacol. 1995, 8, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Shady, A.A.; Colby, B.R.; Cunha, L.F.; Astrin, K.H.; Bishop, D.F.; Desnick, R.J. Congenital erythropoietic porphyria: Identification and expression of eight novel mutationsin the uroporphyrinogen III synthase gene. Br. J. Haematol. 2002, 117, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Warner, C.A.; Desnick, R.J. Congenital erythropoietic porphyria: Identification and expression of 10 mutations in the uroporphyrinogen III synthase gene. J. Clin. Investig. 1995, 95, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Fontanellas, A.; Bensidhoum, M.; Enriquez de Salamanca, R.; Moruno Tirado, A.; de Verneuil, H.; Ged, C. A systematic analysis of the mutations of the uroporphyrinogen III synthase gene in congenital erythropoietic porphyria. Eur. J. Hum. Genet. 1996, 4, 274. [Google Scholar] [PubMed]
- Meguro, K.; Fujita, H.; Ishida, N.; Akagi, R.; Kurihara, T.; Galbraith, R.A.; Kappas, A.; Zabriskie, J.B.; Toback, A.C.; Harber, L.C. Molecular defects of uroporphyrinogen decarboxylase in a patient with mild hepatoerythropoietic porphyria. J. Investig Dermatol. 1994, 102, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Desnick, R.J. The porphyrias: Advances in diagnosis and treatment. Blood 2012, 120, 4496–4504. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Khanna, N.; Kumar, L. Bone marrow transplantation improves symptoms of congenital erythropoietic porphyria even when done post puberty. Indian J. Dermatol. Venereol. Leprol. 2012, 78, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Martinez Peinado, C.; Díaz de Heredia, C.; To-Figueras, J.; Arias-Santiago, S.; Nogueras, P.; Elorza, I.; Olivé, T.; Bádenas, C.; Moreno, M.J.; Tercedor, J.; et al. Successful treatment of congenital erythropoietic porphyria using matched unrelated hematopoietic stem cell transplantation. Pediatr. Dermatol. 2013, 30, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Whatley, S.D.; Ducamp, S.; Gouya, L.; Grandchamp, B.; Beaumont, C.; Badminton, M.N.; Elder, G.H.; Holme, S.A.; Anstey, A.V.; Parker, M.; et al. C-terminal deletions in the q ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am. J. Hum. Genet. 2008, 83, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Magnus, I.A.; Jarrett, A.; Prankerd, T.A.; Rimington, C. Erythropoietic protoporphyria. A new porphyria syndrome with solar urticaria due to protoporphyrinaemia. Lancet 1961, 2, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Harms, J.; Lautenschlager, S.; Minder, C.E.; Minder, E.I. An alpha-melanocyte-stimulating hormone analogue in erythropoietic protoporphyria. N. Engl. J. Med. 2009, 360, 306–307. [Google Scholar] [CrossRef] [PubMed]
- Harms, J.H.; Lautenschlager, S.; Minder, C.E.; Minder, E.I. Mitigating photosensitivity of erythropoietic protoporphyria patients by an agonistic analog of alpha-melanocyte stimulating hormone. Photochem. Photobiol. 2009, 85, 1434–1439. [Google Scholar] [CrossRef] [PubMed]
- Minder, E.I. Afamelanotide, an agonistic analog of alpha-melanocyte-stimulating hormone, in dermal phototoxicity of erythropoietic protoporphyria. Expert Opin. Investig. Drugs 2010, 19, 1591–602. [Google Scholar] [CrossRef] [PubMed]
- Pirlich, M.; Lochs, H.; Schmidt, H.H. Liver cirrhosis in erythropoietic protoporphyria: Improvement of liver function with ursodeoxycholic acid. Am. J. Gastroenterol. 2001, 96, 3468–3469. [Google Scholar] [CrossRef] [PubMed]
- Abitbol, M.; Puy, H.; Sabaté, J.M.; Guénet, J.L.; Deybach, J.C.; Montagutelli, X. Ursodesoxycholic acid and heme-arginate are unable to improve hematopoiesis and liver injury in an erythropoietic protoporphyria mouse model. Physiol. Res. 2006, 55 (Suppl. S2), S93–S101. [Google Scholar] [PubMed]
- Crooks, D.R.; Ghosh, M.C.; Haller, R.G.; Tong, W.H.; Rouault, T.A. Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery. Blood 2010, 115, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.P.; Meek, E.M. Clinical and biochemical improvement following low-dose intravenous iron therapy in a patient with erythropoietic protoporphyria. Br. J. Haematol. 2013, 163, 289–2291. [Google Scholar] [PubMed]
- Milligan, A.; Graham-Brown, R.A.; Sarkany, I.; Baker, H. Erythropoietic protoporphyria exacerbated by oral iron therapy. Br. J. Dermatol. 1988, 119, 63–66. [Google Scholar] [CrossRef] [PubMed]
- McClements, B.M.; Bingham, A.; Callender, M.E.; Trimble, E.R. Erythropoietic protoporphyria and iron therapy. Br. J. Dermatol. 1990, 122, 423–424. [Google Scholar] [CrossRef] [PubMed]
- McGuire, B.M.; Bonkovsky, H.L.; Carithers, R.L., Jr.; Chung, R.T.; Goldstein, L.I.; Lake, J.R.; Lok, A.S.; Potter, C.J.; Rand, E.; Voigt, M.D.; et al. Liver transplantation for erythropoietic protoporphyria liver disease. Liver Transpl. 2005, 11, 1590–1596. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Besur, S.; Hou, W.; Schmeltzer, P.; Bonkovsky, H.L. Clinically Important Features of Porphyrin and Heme Metabolism and the Porphyrias. Metabolites 2014, 4, 977-1006. https://doi.org/10.3390/metabo4040977
Besur S, Hou W, Schmeltzer P, Bonkovsky HL. Clinically Important Features of Porphyrin and Heme Metabolism and the Porphyrias. Metabolites. 2014; 4(4):977-1006. https://doi.org/10.3390/metabo4040977
Chicago/Turabian StyleBesur, Siddesh, Wehong Hou, Paul Schmeltzer, and Herbert L. Bonkovsky. 2014. "Clinically Important Features of Porphyrin and Heme Metabolism and the Porphyrias" Metabolites 4, no. 4: 977-1006. https://doi.org/10.3390/metabo4040977
APA StyleBesur, S., Hou, W., Schmeltzer, P., & Bonkovsky, H. L. (2014). Clinically Important Features of Porphyrin and Heme Metabolism and the Porphyrias. Metabolites, 4(4), 977-1006. https://doi.org/10.3390/metabo4040977