Unraveling Biochemical Pathways Affected by Mitochondrial Dysfunctions Using Metabolomic Approaches



Abstract
:1. Introduction
2. Mitochondrial Structure and Organization
3. Mitochondria and Metabolism
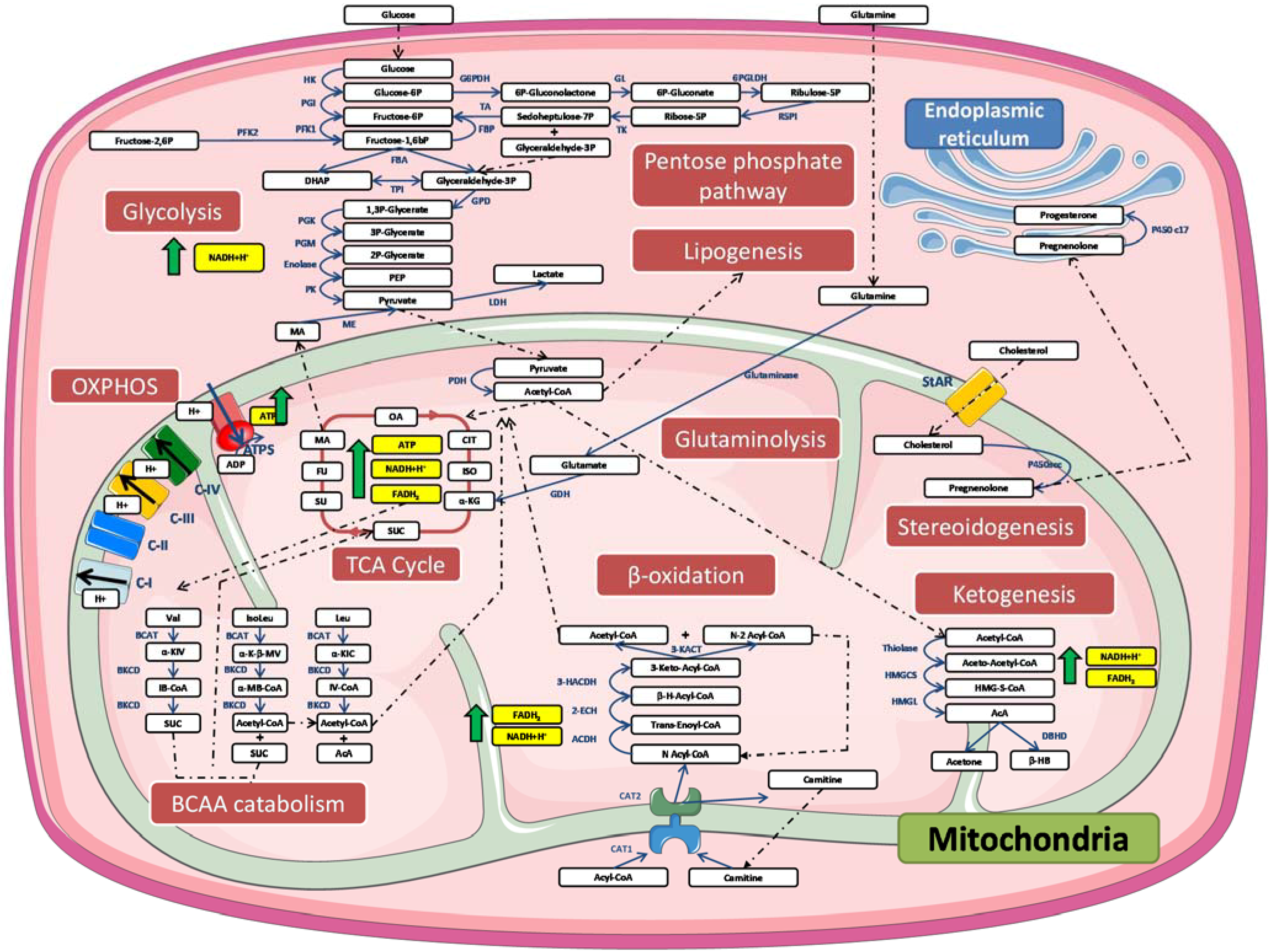
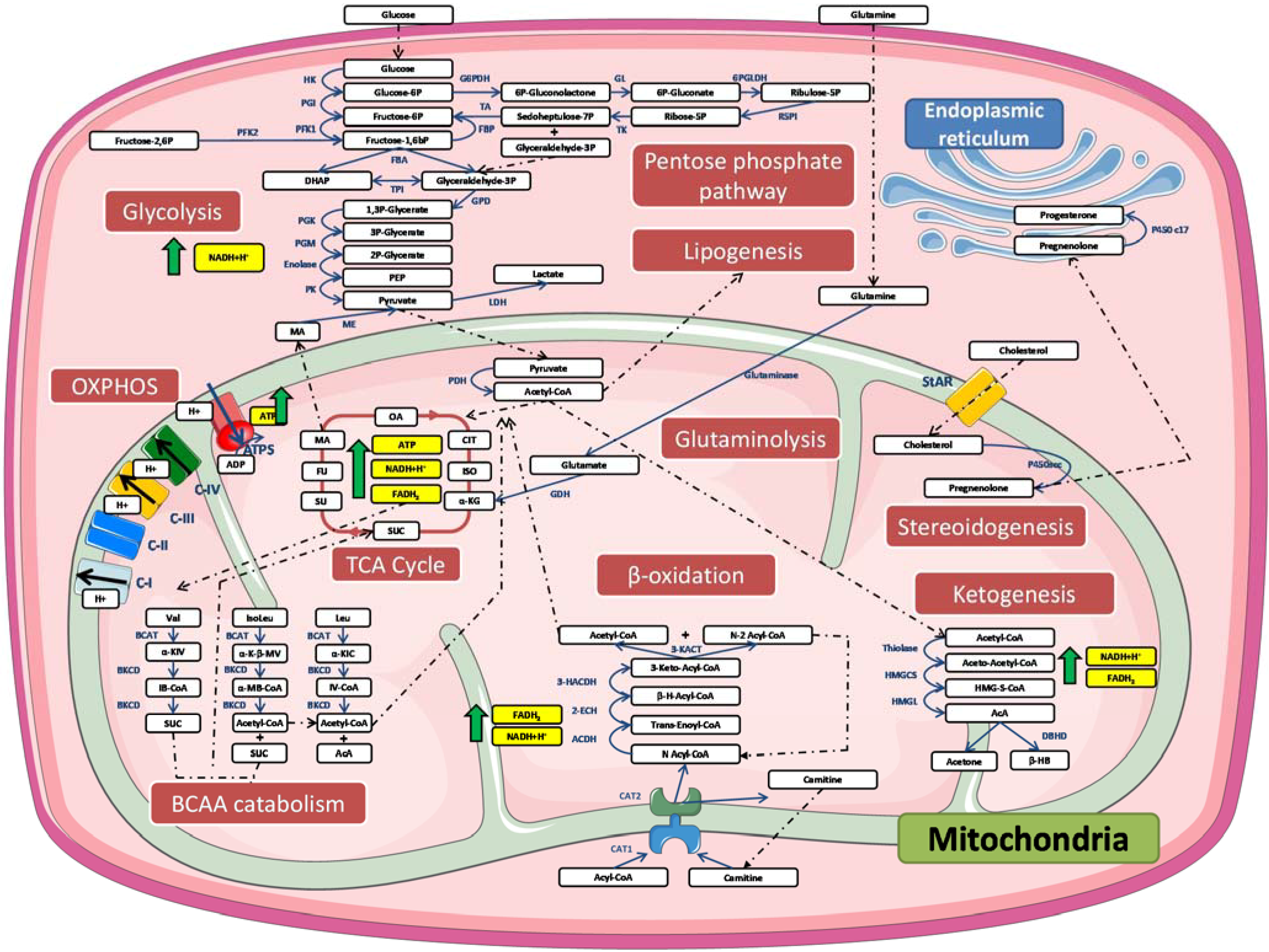
4. Mitochondrial Dysfunction
4.1. Different Types of Mitochondrial Dysfunction
4.2. Mitochondrial Uncoupling and UCP Protein Family

{kind=link}
{kind=link}
| Localization | Function(s) proposed | Species | Tissue or cell-type | Reference | |
|---|---|---|---|---|---|
| UCP-1 | Brown adipocytes, Beige adipocytes, Skeletal muscle | ROS production limitation | Mouse | Skeletal muscle cells | [82] |
| Mitochondrial uncoupling | Mouse | Brown adipose tissue | [83] | ||
| Thermogenesis | Mouse | White adipose tissue | [84] | ||
| UCP-2 | Spleen, Kupfer cells, Immune cells, Central nervous system, Pancreas, Heart, Brown adipocytes | Mitochondrial uncoupling | Yeast | / | [74] |
| ROS production limitation | Mouse | RAW264.7 macrophages | [85] | ||
| Mouse | Endothelial cells | [86] | |||
| Human | Pancreatic adenocarcinoma cell lines | [87] | |||
| Malate, oxaloacetate and aspartate transporter | Human | HepG2 hepatocarcinoma cells | [88] | ||
| UCP-3 | Skeletal muscle, Pancreatic islet beta cells | Mitochondrial uncoupling | Mouse | Skeletal muscle | [89] |
| Fatty acid anion transport (hypothetical) | / | / | [90] | ||
| Ca2+ ion transport | Human | EA.hy926 endothelial cells | [91] | ||
| UCP-4 | Central nervous system | Limited proton translocation | / | Artificial liposomes | [92] |
| Chloride ion transport | / | Artificial liposomes | [92] | ||
| Mitochondrial uncoupling with increased ROS production | Mouse | 3T3-L1 white adipocytes | [93] | ||
| Limitation of ROS production | Human | SH-SY5Y neuroblastoma cells | [94] | ||
| Regulation of mitochondrial complex II activity | Human | SH-SY5Y neuroblastoma cells | [94] | ||
| C. Elegans | / | [95] | |||
| UCP-5 | Central nervous system, Liver, Skeletal muscle | Limited proton translocation | / | Artificial liposomes | [92] |
| Chloride ion transport | / | Artificial liposomes | [92] | ||
| Limitation of ROS production | Human | SH-SY5Y neuroblastoma cells | [94] | ||
| Regulation of mitochondrial complex II activity | Human | SH-SY5Y neuroblastoma cells | [94] |
5. Mitochondrial Uncoupling Proteins and Metabolites Crosstalk
5.1. Glucose
5.2. Nucleotides
5.3. Amino Acids
5.4. Lipids
5.5. Ketone Bodies
5.6. Effect of Plant Metabolites on Mitochondria
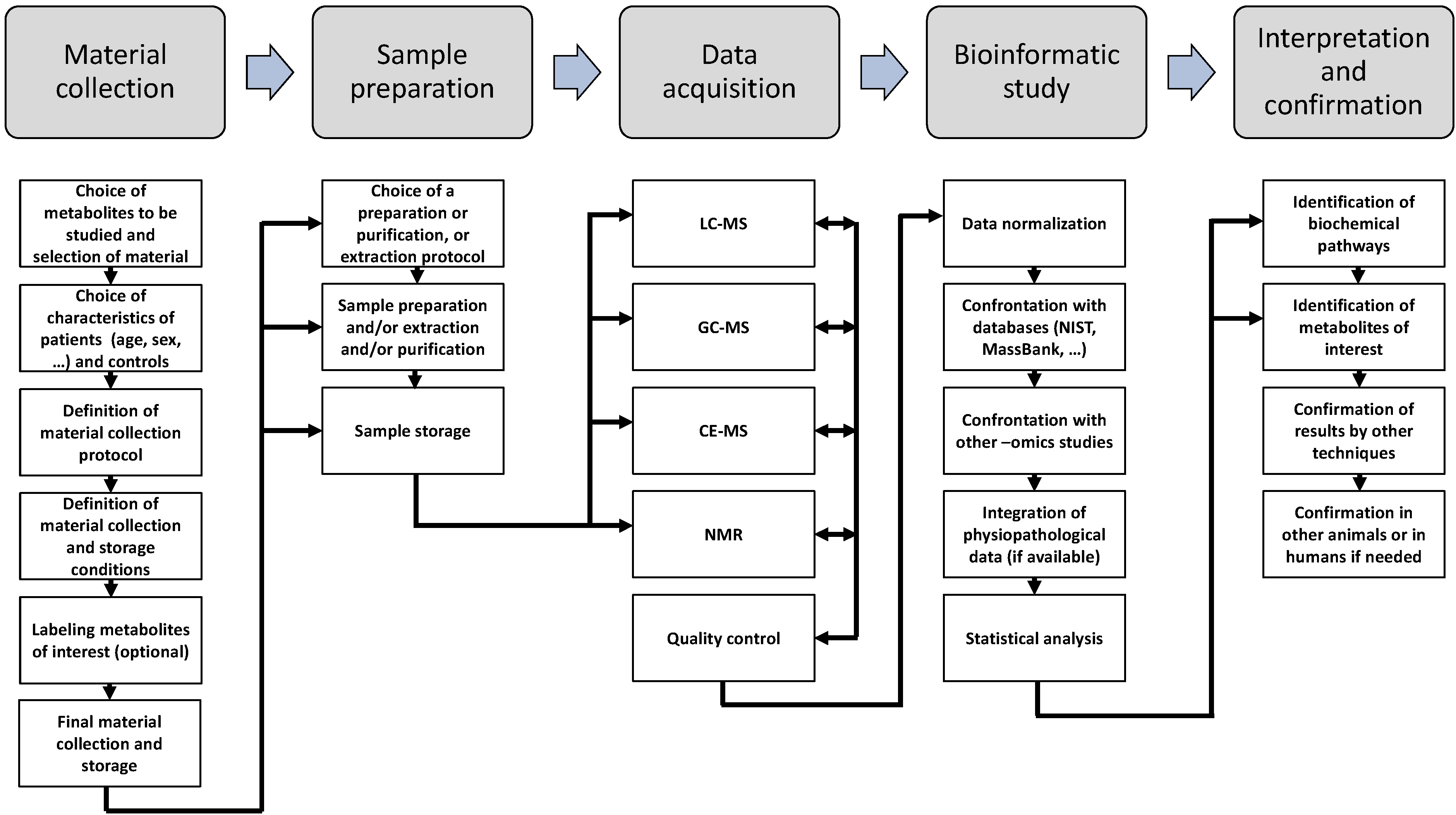
6. Metabolomic Techniquest
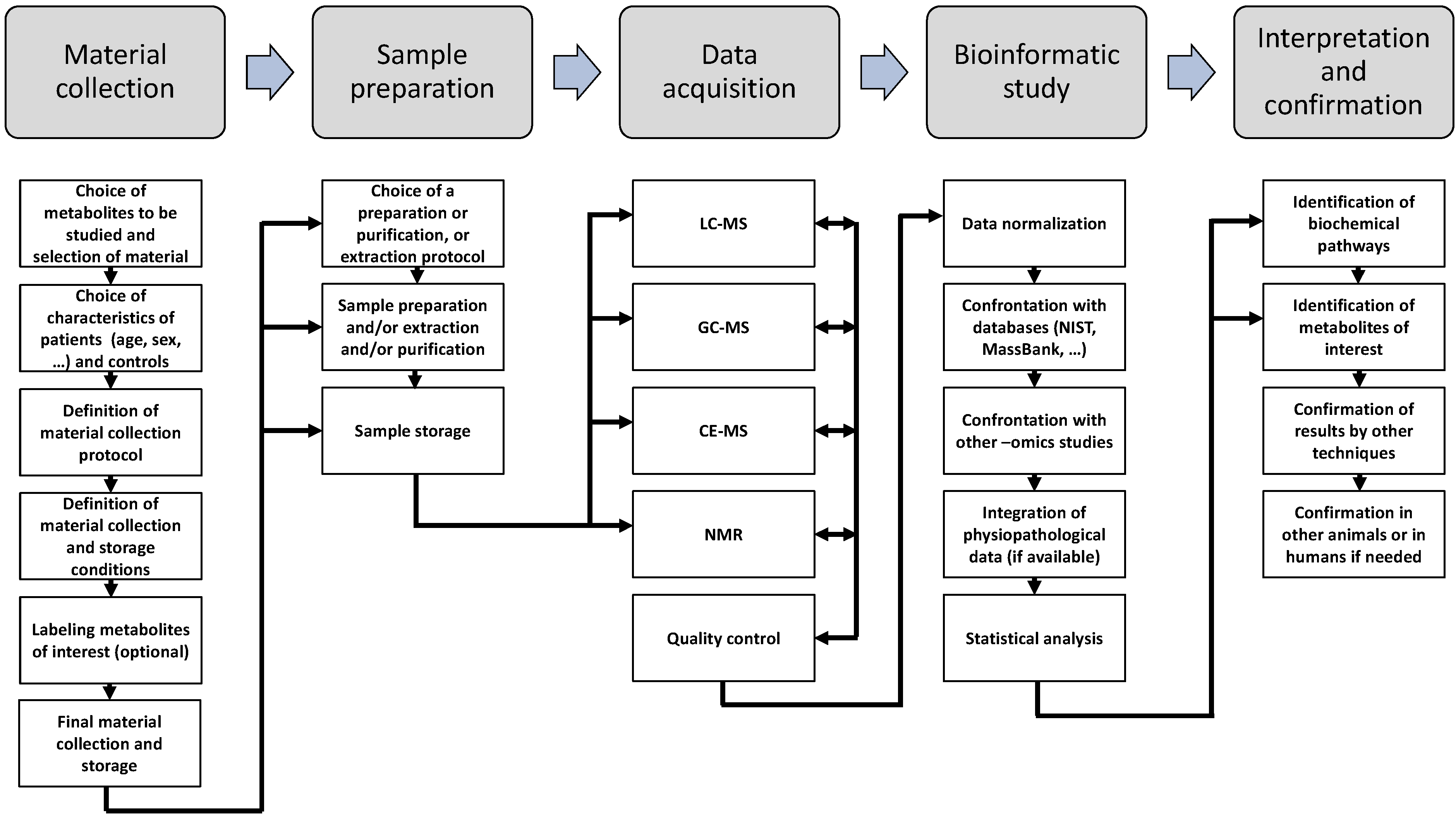
6.1. NMR Spectroscopy
6.2. Mass Spectrometry
7. Impact of Metabolomics in the Field of Obesity, Type II Diabetes Mellitus and Metabolic Syndrome
7.1. Adipose Tissues and Obesity
7.2. Mitochondrial Dysfunction(s) in Obesity and Related Diseases
| Class of metabolites | Metabolite(s) identified | Trend in insulin resistance | Species | Tissue or cell-type | Reference |
|---|---|---|---|---|---|
| Free fatty acids | Myristate, palmitate, stearate, oleate, linoleate, arachidonate | Increase | Human | Blood | [231] |
| 3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid | Increase | Human | Serum | [232] | |
| Carnitines | Dodecenoyl carnitine, tiglyl carnitine, tetradecenoyl carnitine, lauroyl carnitine, propionyl carnitine | Increase | Mice | Blood | [233] |
| Acetylcarnitine, proprionylcarnitine, deoxycarnitine | Increase | Mice | Blood | [234] | |
| Proprionyl carnitine, isovaleryl/2-methylbutyryl carnitine, hexanoyl carnitine, octenoyl carnitine | Increase | Human | Blood | [231] | |
| Carnitine | Increase | Mouse | Serum | [235] | |
| LysoLyso-glycerophospholipids | Lysophosphatidylethanolamine (20:1, 20:2, 22:4) Lysophosphocholine (20:5, 18:2, 18:1) Lysophosphatidyl inositol (20:4) Lysophisphatidylserine (20:0) lysophosphatidic acid (18:2) | Increase | Human | Blood | [236] |
| Lysophosphocholine (16:1) | Decrease | Mice | Blood | [237] | |
| Lysophosphocholine (22:4) | Increase | Mice | Blood | [237] | |
| Lysophosphocholine | Increase | Human | Serum | [238] | |
| Diacyl-phosphatidylcholines Lysophosphocholines | Decrease | Mouse | Serum | [239] | |
| Lysophosphatidylcholine Phosphatidylserine | No change | Mouse | Liver | [237] | |
| Lysophosphatidylcholine (18:0) | Increase | Mouse | Serum | [240] | |
| Amino acids | Serine, glycine, arginine | Decrease | Mouse | Serum | [239] |
| Alanine, arginine, glycine, isoleucine, methionine, ornithine, serine | Decrease | Mouse | Serum | [235] | |
| Valine, leucine, isoleucine, phenylalanine, tyrosine, glutamate/glutamine, aspartate/asparagine, arginine | Increase | Human | Serum | [231] | |
| Citrulline, histidine, methionine, ornithine, proline, serine, tryptophan | No change | Human | Serum | [231] | |
| TCA cycle | Succinate | Increase | Mouse | Serum | [235] |
| Citrate, glucoxe, glycolate, lactate, pyruvate | Decrease | Mouse | Serum | [235] | |
| Ketone bodies | Acetoacetate, acetoneµ | Decrease | Mouse | Serum | [235] |
| 2-hydroxybutyric acid | Increase | Human | Serum | [169,231] | |
| Others | Hydroxysphingomyelin | Decrease | Mouse | Serum | [239] |
7.3. Metabolomic and Lipidomic Studies
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Beger, R.D. A review of applications of metabolomics in cancer. Metabolites 2013, 3, 552–574. [Google Scholar]
- Fiehn, O. Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Comp. Funct. Genom. 2001, 2, 155–168. [Google Scholar]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. “Metabonomics”: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological nmr spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar]
- Robertson, D.G. Metabonomics in toxicology: A review. Toxicol. Sci. 2005, 85, 809–822. [Google Scholar]
- Liesenfeld, D.B.; Habermann, N.; Owen, R.W.; Scalbert, A.; Ulrich, C.M. Review of mass spectrometry-based metabolomics in cancer research. Canc. Epidemiol. Biomarkers Prev. 2013, 22, 2182–2201. [Google Scholar]
- Vermeersch, K.A.; Styczynski, M.P. Applications of metabolomics in cancer research. J. Carcinog. 2013. [Google Scholar] [CrossRef]
- Gray, M.W.; Burger, G.; Lang, B.F. The origin and early evolution of mitochondria. Genome Biol. 2001, 2. Reviews 1018. [Google Scholar]
- Acin-Perez, R.; Fernandez-Silva, P.; Peleato, M.L.; Perez-Martos, A.; Enriquez, J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar]
- Schagger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar]
- Schagger, H.; Pfeiffer, K. The ratio of oxidative phosphorylation complexes i-v in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J. Biol. Chem. 2001, 276, 37861–37867. [Google Scholar]
- Strogolova, V.; Furness, A.; Robb-McGrath, M.; Garlich, J.; Stuart, R.A. Rcf1 and rcf2, members of the hypoxia-induced gene 1 protein family, are critical components of the mitochondrial cytochrome bc1-cytochrome c oxidase supercomplex. Mol. Cell. Biol. 2012, 32, 1363–1373. [Google Scholar]
- Ikeda, K.; Shiba, S.; Horie-Inoue, K.; Shimokata, K.; Inoue, S. A stabilizing factor for mitochondrial respiratory supercomplex assembly regulates energy metabolism in muscle. Nat. Commun. 2013, 4. Article 2147. [Google Scholar]
- Wenz, T.; Hielscher, R.; Hellwig, P.; Schagger, H.; Richers, S.; Hunte, C. Role of phospholipids in respiratory cytochrome bc (1) complex catalysis and supercomplex formation. Biochimica Biophys. Acta 2009, 1787, 609–616. [Google Scholar]
- Bottinger, L.; Horvath, S.E.; Kleinschroth, T.; Hunte, C.; Daum, G.; Pfanner, N.; Becker, T. Phosphatidylethanolamine and cardiolipin differentially affect the stability of mitochondrial respiratory chain supercomplexes. J. Mol. Biol. 2012, 423, 677–686. [Google Scholar]
- Bazan, S.; Mileykovskaya, E.; Mallampalli, V.K.; Heacock, P.; Sparagna, G.C.; Dowhan, W. Cardiolipin-dependent reconstitution of respiratory supercomplexes from purified saccharomyces cerevisiae complexes iii and iv. J. Biol. Chem. 2013, 288, 401–411. [Google Scholar]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, 551–560. [Google Scholar]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar]
- Kubli, D.A.; Gustafsson, A.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar]
- Rizzuto, R.; de Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev.Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar]
- Williams, G.S.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486. [Google Scholar]
- Tait, S.W.; Green, D.R. Mitochondria and cell signalling. J. Cell Sci. 2012, 125, 807–815. [Google Scholar]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar]
- Cardaci, S.; Ciriolo, M.R. Tca cycle defects and cancer: When metabolism tunes redox state. Int. J. Cell Biol. 2012, 2012. Article 161837. [Google Scholar]
- Lazarow, P.B. Rat liver peroxisomes catalyze the beta oxidation of fatty acids. J. Biol. Chem. 1978, 253, 1522–1528. [Google Scholar]
- Foster, D.W. The role of the carnitine system in human metabolism. Ann. NY Acad. Sci. 2004, 1033, 1–16. [Google Scholar]
- Bartlett, K.; Eaton, S. Mitochondrial beta-oxidation. Eur. J. Biochem. 2004, 271, 462–469. [Google Scholar]
- Cho, S.Y.; Kim, J.H.; Paik, Y.K. Cholesterol biosynthesis from lanosterol: Differential inhibition of sterol delta 8-isomerase and other lanosterol-converting enzymes by tamoxifen. Mol. Cells 1998, 8, 233–239. [Google Scholar]
- Cotter, D.G.; Schugar, R.C.; Crawford, P.A. Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, 1060–1076. [Google Scholar]
- Meng, M.; Chen, S.; Lao, T.; Liang, D.; Sang, N. Nitrogen anabolism underlies the importance of glutaminolysis in proliferating cells. Cell Cycle 2010, 9, 3921–3932. [Google Scholar]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mtor and autophagy. Cell 2009, 136, 521–534. [Google Scholar]
- Smith, D.D., Jr.; Campbell, J.W. Distribution of glutamine synthetase and carbamoyl-phosphate synthetase i in vertebrate liver. Proc. Natl. Acad. Sci. USA 1988, 85, 160–164. [Google Scholar]
- Boza, J.J.; Moennoz, D.; Bournot, C.E.; Blum, S.; Zbinden, I.; Finot, P.A.; Ballevre, O. Role of glutamine on the de novo purine nucleotide synthesis in caco-2 cells. Eur. J. Nutr. 2000, 39, 38–46. [Google Scholar]
- Cory, J.G.; Cory, A.H. Critical roles of glutamine as nitrogen donors in purine and pyrimidine nucleotide synthesis: Asparaginase treatment in childhood acute lymphoblastic leukemia. In Vivo 2006, 20, 587–589. [Google Scholar]
- Mouilleron, S.; Badet-Denisot, M.A.; Golinelli-Pimpaneau, B. Glutamine binding opens the ammonia channel and activates glucosamine-6p synthase. J. Biol. Chem. 2006, 281, 4404–4412. [Google Scholar]
- Dang, C.V. Glutaminolysis: Supplying carbon or nitrogen or both for cancer cells? Cell Cycle 2010, 9, 3884–3886. [Google Scholar]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by idh1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar]
- Brosnan, J.T.; Brosnan, M.E. Branched-chain amino acids: Enzyme and substrate regulation. J. Nutr. 2006, 136, 207S–211S. [Google Scholar]
- Elia, M.; Livesey, G. Effects of ingested steak and infused leucine on forelimb metabolism in man and the fate of the carbon skeletons and amino groups of branched-chain amino acids. Clin. Sci. 1983, 64, 517–526. [Google Scholar]
- Suryawan, A.; Hawes, J.W.; Harris, R.A.; Shimomura, Y.; Jenkins, A.E.; Hutson, S.M. A molecular model of human branched-chain amino acid metabolism. Am. J. Clin. Nutr. 1998, 68, 72–81. [Google Scholar]
- Tajiri, K.; Shimizu, Y. Branched-chain amino acids in liver diseases. World J. Gastroenterol. 2013, 19, 7620–7629. [Google Scholar]
- D’Antona, G.; Ragni, M.; Cardile, A.; Tedesco, L.; Dossena, M.; Bruttini, F.; Caliaro, F.; Corsetti, G.; Bottinelli, R.; Carruba, M.O.; et al. Branched-chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle-aged mice. Cell Metabol. 2010, 12, 362–372. [Google Scholar]
- Strum, J.C.; Shehee, R.; Virley, D.; Richardson, J.; Mattie, M.; Selley, P.; Ghosh, S.; Nock, C.; Saunders, A.; Roses, A. Rosiglitazone induces mitochondrial biogenesis in mouse brain. J. Alzheim. Dis. 2007, 11, 45–51. [Google Scholar]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by ppar gamma 2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar]
- Ichikawa, K.; Okabayashi, T.; Shima, Y.; Iiyama, T.; Takezaki, Y.; Munekage, M.; Namikawa, T.; Sugimoto, T.; Kobayashi, M.; Mimura, T.; et al. Branched-chain amino acid-enriched nutrients stimulate antioxidant DNA repair in a rat model of liver injury induced by carbon tetrachloride. Mol. Biol. Rep. 2012, 39, 10803–10810. [Google Scholar]
- Blomstrand, E.; Eliasson, J.; Karlsson, H.K.; Kohnke, R. Branched-chain amino acids activate key enzymes in protein synthesis after physical exercise. J. Nutr. 2006, 136, 269S–273S. [Google Scholar]
- Nishitani, S.; Matsumura, T.; Fujitani, S.; Sonaka, I.; Miura, Y.; Yagasaki, K. Leucine promotes glucose uptake in skeletal muscles of rats. Biochem. Biophys. Res. Comm. 2002, 299, 693–696. [Google Scholar]
- Nishitani, S.; Takehana, K.; Fujitani, S.; Sonaka, I. Branched-chain amino acids improve glucose metabolism in rats with liver cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1292–G1300. [Google Scholar]
- Hinault, C.; Mothe-Satney, I.; Gautier, N.; Lawrence, J.C., Jr.; van Obberghen, E. Amino acids and leucine allow insulin activation of the pkb/mtor pathway in normal adipocytes treated with wortmannin and in adipocytes from db/db mice. FASEB J. 2004, 18, 1894–1896. [Google Scholar]
- Miller, W.L.; Bose, H.S. Early steps in steroidogenesis: Intracellular cholesterol trafficking. J. Lipid Res. 2011, 52, 2111–2135. [Google Scholar]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [Google Scholar]
- Jiang, W.; Wang, S.; Xiao, M.; Lin, Y.; Zhou, L.; Lei, Q.; Xiong, Y.; Guan, K.L.; Zhao, S. Acetylation regulates gluconeogenesis by promoting pepck1 degradation via recruiting the ubr5 ubiquitin ligase. Mol. Cell 2011, 43, 33–44. [Google Scholar]
- Xiong, Y.; Guan, K.L. Mechanistic insights into the regulation of metabolic enzymes by acetylation. J. Cell Biol. 2012, 198, 155–164. [Google Scholar]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar]
- Tuppen, H.A.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 2010, 1797, 113–128. [Google Scholar]
- Park, C.B.; Larsson, N.G. Mitochondrial DNA mutations in disease and aging. J. Cell Biol. 2011, 193, 809–818. [Google Scholar]
- Dudkina, N.V.; Kouril, R.; Peters, K.; Braun, H.P.; Boekema, E.J. Structure and function of mitochondrial supercomplexes. Biochim. Biophys. Acta 2010, 1797, 664–670. [Google Scholar]
- Vannuvel, K.; Renard, P.; Raes, M.; Arnould, T. Functional and morphological impact of er stress on mitochondria. J. Cell. Physiol. 2013, 228, 1802–1818. [Google Scholar]
- Pellegrino, M.W.; Nargund, A.M.; Haynes, C.M. Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 2013, 1833, 410–416. [Google Scholar]
- Rousset, S.; Alves-Guerra, M.C.; Mozo, J.; Miroux, B.; Cassard-Doulcier, A.M.; Bouillaud, F.; Ricquier, D. The biology of mitochondrial uncoupling proteins. Diabetes 2004, 53, S130–S135. [Google Scholar]
- Ricquier, D.; Bouillaud, F.; Toumelin, P.; Mory, G.; Bazin, R.; Arch, J.; Penicaud, L. Expression of uncoupling protein mrna in thermogenic or weakly thermogenic brown adipose tissue. Evidence for a rapid beta-adrenoreceptor-mediated and transcriptionally regulated step during activation of thermogenesis. J. Biol. Chem. 1986, 261, 13905–13910. [Google Scholar]
- Levy, S.E.; Chen, Y.S.; Graham, B.H.; Wallace, D.C. Expression and sequence analysis of the mouse adenine nucleotide translocase 1 and 2 genes. Gene 2000, 254, 57–66. [Google Scholar]
- Stepien, G.; Torroni, A.; Chung, A.B.; Hodge, J.A.; Wallace, D.C. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J. Biol.Chem. 1992, 267, 14592–14597. [Google Scholar]
- Brower, J.V.; Rodic, N.; Seki, T.; Jorgensen, M.; Fliess, N.; Yachnis, A.T.; McCarrey, J.R.; Oh, S.P.; Terada, N. Evolutionarily conserved mammalian adenine nucleotide translocase 4 is essential for spermatogenesis. J. Biol. Chem. 2007, 282, 29658–29666. [Google Scholar]
- Brand, M.D.; Pakay, J.L.; Ocloo, A.; Kokoszka, J.; Wallace, D.C.; Brookes, P.S.; Cornwall, E.J. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem. J. 2005, 392, 353–362. [Google Scholar]
- Liu, Y.; Chen, X.J. Adenine nucleotide translocase, mitochondrial stress, and degenerative cell death. Oxidative Med. Cell. Longev. 2013, 2013. Article 146860. [Google Scholar]
- Liu, J.; Li, J.; Li, W.J.; Wang, C.M. The role of uncoupling proteins in diabetes mellitus. J. Diabetes Res. 2013, 2013. Article 585897. [Google Scholar]
- Klingenberg, M.; Winkler, E. The reconstituted isolated uncoupling protein is a membrane potential driven h+ translocator. EMBO J. 1985, 4, 3087–3092. [Google Scholar]
- Matthias, A.; Ohlson, K.B.; Fredriksson, J.M.; Jacobsson, A.; Nedergaard, J.; Cannon, B. Thermogenic responses in brown fat cells are fully ucp1-dependent. Ucp2 or ucp3 do not substitute for ucp1 in adrenergically or fatty scid-induced thermogenesis. J. Biol. Chem. 2000, 275, 25073–25081. [Google Scholar]
- Cadrin, M.; Tolszczuk, M.; Guy, J.; Pelletier, G.; Freeman, K.B.; Bukowiecki, L.J. Immunohistochemical identification of the uncoupling protein in rat brown adipose tissue. J. Histochem. Cytochem. 1985, 33, 150–154. [Google Scholar]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor gamma (ppargamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, ucp1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar]
- Boss, O.; Samec, S.; Paoloni-Giacobino, A.; Rossier, C.; Dulloo, A.; Seydoux, J.; Muzzin, P.; Giacobino, J.P. Uncoupling protein-3: A new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett. 1997, 408, 39–42. [Google Scholar]
- Sale, M.M.; Hsu, F.C.; Palmer, N.D.; Gordon, C.J.; Keene, K.L.; Borgerink, H.M.; Sharma, A.J.; Bergman, R.N.; Taylor, K.D.; Saad, M.F.; et al. The uncoupling protein 1 gene, ucp1, is expressed in mammalian islet cells and associated with acute insulin response to glucose in african american families from the iras family study. BMC Endocr. Disord. 2007, 7, 1. [Google Scholar]
- Chen, Y.; Li, Z.Y.; Yang, Y.; Zhang, H.J. Uncoupling protein 2 regulates glucagon-like peptide-1 secretion in l-cells. World J. Gastroenterol. 2012, 18, 3451–3457. [Google Scholar]
- Fleury, C.; Neverova, M.; Collins, S.; Raimbault, S.; Champigny, O.; Levi-Meyrueis, C.; Bouillaud, F.; Seldin, M.F.; Surwit, R.S.; Ricquier, D.; et al. Uncoupling protein-2: A novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 1997, 15, 269–272. [Google Scholar]
- Boss, O.; Samec, S.; Dulloo, A.; Seydoux, J.; Muzzin, P.; Giacobino, J.P. Tissue-dependent upregulation of rat uncoupling protein-2 expression in response to fasting or cold. FEBS Lett. 1997, 412, 111–114. [Google Scholar]
- Li, W.; Nichols, K.; Nathan, C.A.; Zhao, Y. Mitochondrial uncoupling protein 2 is up-regulated in human head and neck, skin, pancreatic, and prostate tumors. Canc. Biomarkers 2013, 13, 377–383. [Google Scholar]
- Li, Y.; Maedler, K.; Shu, L.; Haataja, L. Ucp-2 and ucp-3 proteins are differentially regulated in pancreatic beta-cells. PloS One 2008. [Google Scholar] [CrossRef]
- Mao, W.; Yu, X.X.; Zhong, A.; Li, W.; Brush, J.; Sherwood, S.W.; Adams, S.H.; Pan, G. Ucp4, a novel brain-specific mitochondrial protein that reduces membrane potential in mammalian cells. FEBS Lett. 1999, 443, 326–330. [Google Scholar]
- Yu, X.X.; Mao, W.; Zhong, A.; Schow, P.; Brush, J.; Sherwood, S.W.; Adams, S.H.; Pan, G. Characterization of novel ucp5/bmcp1 isoforms and differential regulation of ucp4 and ucp5 expression through dietary or temperature manipulation. FASEB J. 2000, 14, 1611–1618. [Google Scholar]
- Bouillaud, F.; Ricquier, D.; Thibault, J.; Weissenbach, J. Molecular approach to thermogenesis in brown adipose tissue: Cdna cloning of the mitochondrial uncoupling protein. Proc. Natl. Acad. of Sci. USA 1985, 82, 445–448. [Google Scholar]
- Kukat, A.; Dogan, S.A.; Edgar, D.; Mourier, A.; Jacoby, C.; Maiti, P.; Mauer, J.; Becker, C.; Senft, K.; Wibom, R.; et al. Loss of ucp2 attenuates mitochondrial dysfunction without altering ros production and uncoupling activity. PLoS Genet. 2014, 10, e1004385. [Google Scholar]
- Adjeitey, C.N.; Mailloux, R.J.; Dekemp, R.A.; Harper, M.E. Mitochondrial uncoupling in skeletal muscle by ucp1 augments energy expenditure and glutathione content while mitigating ros production. Am. J. Physiol. Endocrinol. Metabol. 2013, 305, E405–E415. [Google Scholar]
- Shabalina, I.G.; Vrbacky, M.; Pecinova, A.; Kalinovich, A.V.; Drahota, Z.; Houstek, J.; Mracek, T.; Cannon, B.; Nedergaard, J. Ros production in brown adipose tissue mitochondria: The question of ucp1-dependence. Biochim. Biophys. Acta 2014. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Petrovic, N.; de Jong, J.M.; Kalinovich, A.V.; Cannon, B.; Nedergaard, J. Ucp1 in brite/beige adipose tissue mitochondria is functionally thermogenic. Cell Rep. 2013, 5, 1196–1203. [Google Scholar]
- Wang, Y.; Huang, L.; Abdelrahim, M.; Cai, Q.; Truong, A.; Bick, R.; Poindexter, B.; Sheikh-Hamad, D. Stanniocalcin-1 suppresses superoxide generation in macrophages through induction of mitochondrial ucp2. J. Leukocyte Biol. 2009, 86, 981–988. [Google Scholar]
- Tian, X.Y.; Wong, W.T.; Xu, A.; Lu, Y.; Zhang, Y.; Wang, L.; Cheang, W.S.; Wang, Y.; Yao, X.; Huang, Y. Uncoupling protein-2 protects endothelial function in diet-induced obese mice. Circ. Res. 2012, 110, 1211–1216. [Google Scholar]
- Dando, I.; Fiorini, C.; Pozza, E.D.; Padroni, C.; Costanzo, C.; Palmieri, M.; Donadelli, M. Ucp2 inhibition triggers ros-dependent nuclear translocation of gapdh and autophagic cell death in pancreatic adenocarcinoma cells. Biochim. Biophys. Acta 2013, 1833, 672–679. [Google Scholar]
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. Ucp2 transports c4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar]
- Clapham, J.C.; Arch, J.R.; Chapman, H.; Haynes, A.; Lister, C.; Moore, G.B.; Piercy, V.; Carter, S.A.; Lehner, I.; Smith, S.A.; et al. Mice overexpressing human uncoupling protein-3 in skeletal muscle are hyperphagic and lean. Nature 2000, 406, 415–418. [Google Scholar]
- Himms-Hagen, J.; Harper, M.E. Physiological role of ucp3 may be export of fatty acids from mitochondria when fatty acid oxidation predominates: An hypothesis. Exp. Biol. Med. 2001, 226, 78–84. [Google Scholar]
- Trenker, M.; Malli, R.; Fertschai, I.; Levak-Frank, S.; Graier, W.F. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat. Cell Biol. 2007, 9, 445–452. [Google Scholar]
- Hoang, T.; Smith, M.D.; Jelokhani-Niaraki, M. Toward understanding the mechanism of ion transport activity of neuronal uncoupling proteins ucp2, ucp4, and ucp5. Biochemistry 2012, 51, 4004–4014. [Google Scholar]
- Gao, C.L.; Zhu, J.G.; Zhao, Y.P.; Chen, X.H.; Ji, C.B.; Zhang, C.M.; Zhu, C.; Xia, Z.K.; Peng, Y.Z.; Guo, X.R. Mitochondrial dysfunction is induced by the overexpression of ucp4 in 3t3-l1 adipocytes. Int. J. Mol. Med. 2010, 25, 71–80. [Google Scholar]
- Ho, J.W.; Ho, P.W.; Zhang, W.Y.; Liu, H.F.; Kwok, K.H.; Yiu, D.C.; Chan, K.H.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Transcriptional regulation of ucp4 by nf-kappab and its role in mediating protection against mpp+ toxicity. Free Radic. Biol. Med. 2010, 49, 192–204. [Google Scholar]
- Pfeiffer, M.; Kayzer, E.B.; Yang, X.; Abramson, E.; Kenaston, M.A.; Lago, C.U.; Lo, H.H.; Sedensky, M.M.; Lunceford, A.; Clarke, C.F.; et al. Caenorhabditis elegans ucp4 protein controls complex ii-mediated oxidative phosphorylation through succinate transport. J. Biol. Chem. 2011, 286, 37712–37720. [Google Scholar]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Sign. 2011, 15, 1583–1606. [Google Scholar]
- Kirkman, H.N.; Gaetani, G.F. Mammalian catalase: A venerable enzyme with new mysteries. Trends Biochem. Sci. 2007, 32, 44–50. [Google Scholar]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar]
- Wood, Z.A.; Schroder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar]
- Lundberg, M.; Johansson, C.; Chandra, J.; Enoksson, M.; Jacobsson, G.; Ljung, J.; Johansson, M.; Holmgren, A. Cloning and expression of a novel human glutaredoxin (grx2) with mitochondrial and nuclear isoforms. J. Biol. Chem. 2001, 276, 26269–26275. [Google Scholar]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschop, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar]
- Cui, Y.; Xu, X.; Bi, H.; Zhu, Q.; Wu, J.; Xia, X.; Qiushi, R.; Ho, P.C. Expression modification of uncoupling proteins and mnsod in retinal endothelial cells and pericytes induced by high glucose: The role of reactive oxygen species in diabetic retinopathy. Exp. Eye Res. 2006, 83, 807–816. [Google Scholar]
- Dymkowska, D.; Drabarek, B.; Podszywalow-Bartnicka, P.; Szczepanowska, J.; Zablocki, K. Hyperglycaemia modifies energy metabolism and reactive oxygen species formation in endothelial cells in vitro. Arch. Biochem. Biophys. 2014, 542, 7–13. [Google Scholar]
- Koziel, A.; Woyda-Ploszczyca, A.; Kicinska, A.; Jarmuszkiewicz, W. The influence of high glucose on the aerobic metabolism of endothelial ea.Hy926 cells. Pflugers Arch. Eur. J. Physiol. 2012, 464, 657–669. [Google Scholar]
- Matsuzaki, H.; Daitoku, H.; Hatta, M.; Aoyama, H.; Yoshimochi, K.; Fukamizu, A. Acetylation of foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. USA 2005, 102, 11278–11283. [Google Scholar]
- Nakae, J.; Cao, Y.; Oki, M.; Orba, Y.; Sawa, H.; Kiyonari, H.; Iskandar, K.; Suga, K.; Lombes, M.; Hayashi, Y. Forkhead transcription factor foxo1 in adipose tissue regulates energy storage and expenditure. Diabetes 2008, 57, 563–576. [Google Scholar]
- Komelina, N.P.; Amerkhanov, Z.G. A comparative study of the inhibitory effects of purine nucleotides and carboxyatractylate on the uncoupling protein-3 and adenine nucleotide translocase. Acta Biochim. Pol. 2010, 57, 413–419. [Google Scholar]
- Winkler, E.; Wachter, E.; Klingenberg, M. Identification of the ph sensor for nucleotide binding in the uncoupling protein from brown adipose tissue. Biochemistry 1997, 36, 148–155. [Google Scholar]
- Winkler, E.; Klingenberg, M. Photoaffinity labeling of the nucleotide-binding site of the uncoupling protein from hamster brown adipose tissue. Eur. J. Biochem. 1992, 203, 295–304. [Google Scholar]
- Arechaga, I.; Ledesma, A.; Rial, E. The mitochondrial uncoupling protein ucp1: A gated pore. IUBMB Life 2001, 52, 165–173. [Google Scholar]
- Du, Y.; Meng, Q.; Zhang, Q.; Guo, F. Isoleucine or valine deprivation stimulates fat loss via increasing energy expenditure and regulating lipid metabolism in wat. Amino Acids 2012, 43, 725–734. [Google Scholar]
- Cheng, Y.; Meng, Q.; Wang, C.; Li, H.; Huang, Z.; Chen, S.; Xiao, F.; Guo, F. Leucine deprivation decreases fat mass by stimulation of lipolysis in white adipose tissue and upregulation of uncoupling protein 1 (ucp1) in brown adipose tissue. Diabetes 2010, 59, 17–25. [Google Scholar]
- Cheng, Y.; Zhang, Q.; Meng, Q.; Xia, T.; Huang, Z.; Wang, C.; Liu, B.; Chen, S.; Xiao, F.; Du, Y.; et al. Leucine deprivation stimulates fat loss via increasing crh expression in the hypothalamus and activating the sympathetic nervous system. Mol. Endocrinol. 2011, 25, 1624–1635. [Google Scholar]
- Bernardi, P.; Penzo, D.; Wojtczak, L. Mitochondrial energy dissipation by fatty acids. Mechanisms and implications for cell death. Vitam. Horm. 2002, 65, 97–126. [Google Scholar]
- Schonfeld, P.; Wojtczak, L. Fatty acids decrease mitochondrial generation of reactive oxygen species at the reverse electron transport but increase it at the forward transport. Biochim. Biophys. Acta 2007, 1767, 1032–1040. [Google Scholar]
- Cole, M.A.; Murray, A.J.; Cochlin, L.E.; Heather, L.C.; McAleese, S.; Knight, N.S.; Sutton, E.; Jamil, A.A.; Parassol, N.; Clarke, K. A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res. Cardiol. 2011, 106, 447–457. [Google Scholar]
- Lambertucci, R.H.; Leandro, C.G.; Vinolo, M.A.; Nachbar, R.T.; Dos Reis Silveira, L.; Hirabara, S.M.; Curi, R.; Pithon-Curi, T.C. The effects of palmitic acid on nitric oxide production by rat skeletal muscle: Mechanism via superoxide and inos activation. Cell. Physiol. Biochem 2012, 30, 1169–1180. [Google Scholar]
- Divakaruni, A.S.; Humphrey, D.M.; Brand, M.D. Fatty acids change the conformation of uncoupling protein 1 (ucp1). J. Biol. Chem. 2012, 287, 36845–36853. [Google Scholar]
- Fedorenko, A.; Lishko, P.V.; Kirichok, Y. Mechanism of fatty-acid-dependent ucp1 uncoupling in brown fat mitochondria. Cell 2012, 151, 400–413. [Google Scholar]
- Beck, V.; Jaburek, M.; Demina, T.; Rupprecht, A.; Porter, R.K.; Jezek, P.; Pohl, E.E. Polyunsaturated fatty acids activate human uncoupling proteins 1 and 2 in planar lipid bilayers. FASEB J. 2007, 21, 1137–1144. [Google Scholar]
- Taltavull, N.; Munoz-Cortes, M.; Lluis, L.; Jove, M.; Fortuno, A.; Molinar-Toribio, E.; Torres, J.L.; Pazos, M.; Medina, I.; Nogues, M.R. Eicosapentaenoic acid/docosahexaenoic acid 1:1 ratio improves histological alterations in obese rats with metabolic syndrome. Lipids Health Dis. 2014. [Google Scholar] [CrossRef]
- Oster, R.T.; Tishinsky, J.M.; Yuan, Z.; Robinson, L.E. Docosahexaenoic acid increases cellular adiponectin mrna and secreted adiponectin protein, as well as ppargamma mrna, in 3t3-l1 adipocytes. Appl. Physiol. Nutr. Metabol. 2010, 35, 783–789. [Google Scholar]
- Sadurskis, A.; Dicker, A.; Cannon, B.; Nedergaard, J. Polyunsaturated fatty acids recruit brown adipose tissue: Increased ucp content and nst capacity. Am. J. Physiol. 1995, 269, E351–E360. [Google Scholar]
- Jeckel, K.M.; Veeramachaneni, D.N.; Chicco, A.J.; Chapman, P.L.; Mulligan, C.M.; Hegarty, J.R.; Pagliassotti, M.J.; Ferguson, L.A.; Bouma, G.J.; Frye, M.A. Docosahexaenoic acid supplementation does not improve western diet-induced cardiomyopathy in rats. PloS One 2012. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, C.; Dong, Y.; Wang, S.; Song, P.; Viollet, B.; Zou, M.H. Activation of the amp-activated protein kinase by eicosapentaenoic acid (epa, 20:5 n-3) improves endothelial function in vivo. PloS One 2012, 7, e35508. [Google Scholar]
- Flachs, P.; Horakova, O.; Brauner, P.; Rossmeisl, M.; Pecina, P.; Franssen-van Hal, N.; Ruzickova, J.; Sponarova, J.; Drahota, Z.; Vlcek, C.; et al. Polyunsaturated fatty acids of marine origin upregulate mitochondrial biogenesis and induce beta-oxidation in white fat. Diabetologia 2005, 48, 2365–2375. [Google Scholar]
- Janovska, P.; Flachs, P.; Kazdova, L.; Kopecky, J. Anti-obesity effect of n-3 polyunsaturated fatty acids in mice fed high-fat diet is independent of cold-induced thermogenesis. Physiol.Res. 2013, 62, 153–161. [Google Scholar]
- Flachs, P.; Ruhl, R.; Hensler, M.; Janovska, P.; Zouhar, P.; Kus, V.; Macek Jilkova, Z.; Papp, E.; Kuda, O.; Svobodova, M.; et al. Synergistic induction of lipid catabolism and anti-inflammatory lipids in white fat of dietary obese mice in response to calorie restriction and n-3 fatty acids. Diabetologia 2011, 54, 2626–2638. [Google Scholar]
- Flachs, P.; Rossmeisl, M.; Bryhn, M.; Kopecky, J. Cellular and molecular effects of n-3 polyunsaturated fatty acids on adipose tissue biology and metabolism. Clin. Sci. 2009, 116, 1–16. [Google Scholar]
- Griffiths, D.E.; Cain, K.; Hyams, R.L. Studies of energy-linked reactions. Inhibition of oxidative phosphorylation by dl-8-methyldihydrolipoate. Biochem. J. 1977, 164, 699–704. [Google Scholar]
- Valdecantos, M.P.; Perez-Matute, P.; Gonzalez-Muniesa, P.; Prieto-Hontoria, P.L.; Moreno-Aliaga, M.J.; Martinez, J.A. Lipoic acid administration prevents nonalcoholic steatosis linked to long-term high-fat feeding by modulating mitochondrial function. J. Nutr. Biochem. 2012, 23, 1676–1684. [Google Scholar]
- Wang, Y.; Li, X.; Guo, Y.; Chan, L.; Guan, X. Alpha-lipoic acid increases energy expenditure by enhancing adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor-gamma coactivator-1alpha signaling in the skeletal muscle of aged mice. Metabol. Clin. Exp. 2010, 59, 967–976. [Google Scholar]
- Khamaisi, M.; Potashnik, R.; Tirosh, A.; Demshchak, E.; Rudich, A.; Tritschler, H.; Wessel, K.; Bashan, N. Lipoic acid reduces glycemia and increases muscle glut4 content in streptozotocin-diabetic rats. Metabol. Clin. Exp. 1997, 46, 763–768. [Google Scholar]
- Rudich, A.; Tirosh, A.; Potashnik, R.; Khamaisi, M.; Bashan, N. Lipoic acid protects against oxidative stress induced impairment in insulin stimulation of protein kinase b and glucose transport in 3t3-l1 adipocytes. Diabetologia 1999, 42, 949–957. [Google Scholar]
- Valdecantos, M.P.; Perez-Matute, P.; Quintero, P.; Martinez, J.A. Vitamin c, resveratrol and lipoic acid actions on isolated rat liver mitochondria: All antioxidants but different. Redox Rep. 2010, 15, 207–216. [Google Scholar]
- Tonin, A.M.; Amaral, A.U.; Busanello, E.N.; Grings, M.; Castilho, R.F.; Wajner, M. Long-chain 3-hydroxy fatty acids accumulating in long-chain 3-hydroxyacyl-coa dehydrogenase and mitochondrial trifunctional protein deficiencies uncouple oxidative phosphorylation in heart mitochondria. J. Bioenerg. Biomembr. 2013, 45, 47–57. [Google Scholar]
- Sarkar, P.; Zaja, I.; Bienengraeber, M.; Rarick, K.R.; Terashvili, M.; Canfield, S.; Falck, J.R.; Harder, D.R. Epoxyeicosatrienoic acids pre-treatment improves amyloid beta-induced mitochondrial dysfunction in cultured rat hippocampal astrocytes. Am. J. Physiol. Heart Circ. Physiol. 2013. [Google Scholar]
- Echtay, K.S.; Esteves, T.C.; Pakay, J.L.; Jekabsons, M.B.; Lambert, A.J.; Portero-Otin, M.; Pamplona, R.; Vidal-Puig, A.J.; Wang, S.; Roebuck, S.J.; et al. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 2003, 22, 4103–4110. [Google Scholar]
- Malingriaux, E.A.; Rupprecht, A.; Gille, L.; Jovanovic, O.; Jezek, P.; Jaburek, M.; Pohl, E.E. Fatty acids are key in 4-hydroxy-2-nonenal-mediated activation of uncoupling proteins 1 and 2. PloS One 2013. [Google Scholar] [CrossRef]
- Nadtochiy, S.M.; Zhu, Q.; Urciuoli, W.; Rafikov, R.; Black, S.M.; Brookes, P.S. Nitroalkenes confer acute cardioprotection via adenine nucleotide translocase 1. J. Biol. Chem. 2012, 287, 3573–3580. [Google Scholar]
- Senese, R.; Valli, V.; Moreno, M.; Lombardi, A.; Busiello, R.A.; Cioffi, F.; Silvestri, E.; Goglia, F.; Lanni, A.; de Lange, P. Uncoupling protein 3 expression levels influence insulin sensitivity, fatty acid oxidation, and related signaling pathways. Pflug. Arch. 2011, 461, 153–164. [Google Scholar]
- Srivastava, S.; Kashiwaya, Y.; King, M.T.; Baxa, U.; Tam, J.; Niu, G.; Chen, X.; Clarke, K.; Veech, R.L. Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet. FASEB J. 2012, 26, 2351–2362. [Google Scholar]
- Kashiwaya, Y.; Pawlosky, R.; Markis, W.; King, M.T.; Bergman, C.; Srivastava, S.; Murray, A.; Clarke, K.; Veech, R.L. A ketone ester diet increases brain malonyl-coa and uncoupling proteins 4 and 5 while decreasing food intake in the normal wistar rat. J. Biol. Chem. 2010, 285, 25950–25956. [Google Scholar]
- Sullivan, P.G.; Rippy, N.A.; Dorenbos, K.; Concepcion, R.C.; Agarwal, A.K.; Rho, J.M. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann. Neurol. 2004, 55, 576–580. [Google Scholar]
- Jikumaru, M.; Hiramoto, K.; Honma, T.; Sato, E.F.; Sekiyama, A.; Inoue, M. Effect of starvation on the survival of male and female mice. Physiol. Chem. Phys. Med. NMR 2007, 39, 247–257. [Google Scholar]
- Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Lamb, R.; Hulit, J.; Howell, A.; Gandara, R.; Sartini, M.; Rubin, E.; Lisanti, M.P.; Sotgia, F. Mitochondrial dysfunction in breast cancer cells prevents tumor growth: Understanding chemoprevention with metformin. Cell Cycle 2013, 12, 172–182. [Google Scholar]
- Mercader, J.; Ribot, J.; Murano, I.; Felipe, F.; Cinti, S.; Bonet, M.L.; Palou, A. Remodeling of white adipose tissue after retinoic acid administration in mice. Endocrinology 2006, 147, 5325–5332. [Google Scholar]
- Amengual, J.; Ribot, J.; Bonet, M.L.; Palou, A. Retinoic acid treatment enhances lipid oxidation and inhibits lipid biosynthesis capacities in the liver of mice. Cell. Physiol. Biochem. 2010, 25, 657–666. [Google Scholar]
- Camara, Y.; Mampel, T.; Armengol, J.; Villarroya, F.; Dejean, L. Ucp3 expression in liver modulates gene expression and oxidative metabolism in response to fatty acids, and sensitizes mitochondria to permeability transition. Cell. Phys. Biochem. 2009, 24, 243–252. [Google Scholar]
- Pietta, P.G. Flavonoids as antioxidants. J. Nat. Prod. 2000, 63, 1035–1042. [Google Scholar]
- Dorta, D.J.; Pigoso, A.A.; Mingatto, F.E.; Rodrigues, T.; Pestana, C.R.; Uyemura, S.A.; Santos, A.C.; Curti, C. Antioxidant activity of flavonoids in isolated mitochondria. Phytother. Res. 2008, 22, 1213–1218. [Google Scholar]
- Miyashita, K.; Nishikawa, S.; Beppu, F.; Tsukui, T.; Abe, M.; Hosokawa, M. The allenic carotenoid fucoxanthin, a novel marine nutraceutical from brown seaweeds. J. Sci. Food Agr. 2011, 91, 1166–1174. [Google Scholar]
- Schuster, S.; Fell, D.A.; Dandekar, T. A general definition of metabolic pathways useful for systematic organization and analysis of complex metabolic networks. Nat. Biotechnol. 2000, 18, 326–332. [Google Scholar]
- Edwards, J.S.; Ibarra, R.U.; Palsson, B.O. In silico predictions of escherichia coli metabolic capabilities are consistent with experimental data. Nat. Biotechnol. 2001, 19, 125–130. [Google Scholar]
- Gowda, G.A.; Zhang, S.; Gu, H.; Asiago, V.; Shanaiah, N.; Raftery, D. Metabolomics-based methods for early disease diagnostics. Expert Rev. Mol. Diagn. 2008, 8, 617–633. [Google Scholar]
- Shulaev, V. Metabolomics technology and bioinformatics. Brief. Bioinform. 2006, 7, 128–139. [Google Scholar]
- Beecher, C.W.W. The human metabolome. In Metabolic Profiling: Its Role in Biomarker Discovery and Gene Function Analysis; Harrigan, G.G., Goodacre, R, Eds.; Springer: Berlin, Germany, 2003. [Google Scholar]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. Hmdb: The human metabolome database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar]
- Weckwerth, W.; Wenzel, K.; Fiehn, O. Process for the integrated extraction, identification and quantification of metabolites, proteins and rna to reveal their co-regulation in biochemical networks. Proteomics 2004, 4, 78–83. [Google Scholar]
- Fan, T.W.; Lorkiewicz, P.K.; Sellers, K.; Moseley, H.N.; Higashi, R.M.; Lane, A.N. Stable isotope-resolved metabolomics and applications for drug development. Pharmacol. Therapeut. 2012, 133, 366–391. [Google Scholar]
- Smolinska, A.; Blanchet, L.; Buydens, L.M.; Wijmenga, S.S. Nmr and pattern recognition methods in metabolomics: From data acquisition to biomarker discovery: A review. Anal. Chim. Acta 2012, 750, 82–97. [Google Scholar]
- Zhou, B.; Xiao, J.F.; Tuli, L.; Ressom, H.W. Lc-ms-based metabolomics. Mol. Biosyst. 2012, 8, 470–481. [Google Scholar]
- Koek, M.M.; Jellema, R.H.; van der Greef, J.; Tas, A.C.; Hankemeier, T. Quantitative metabolomics based on gas chromatography mass spectrometry: Status and perspectives. Metabolomics 2011, 7, 307–328. [Google Scholar]
- Werner, E.; Heilier, J.F.; Ducruix, C.; Ezan, E.; Junot, C.; Tabet, J.C. Mass spectrometry for the identification of the discriminating signals from metabolomics: Current status and future trends. J. Chromatogr. B 2008, 871, 143–163. [Google Scholar]
- Bakken, I.J.; Sonnewald, U.; Clark, J.B.; Bates, T.E. [U-13C]glutamate metabolism in rat brain mitochondria reveals malic enzyme activity. Neuroreport 1997, 8, 1567–1570. [Google Scholar]
- Teng, Q.; Ekman, D.R.; Huang, W.; Collette, T.W. Push-through direct injection nmr: An optimized automation method applied to metabolomics. Analyst 2012, 137, 2226–2232. [Google Scholar]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Matrix effect in quantitative lc/ms/ms analyses of biological fluids: A method for determination of finasteride in human plasma at picogram per milliliter concentrations. Anal. Chem. 1998, 70, 882–889. [Google Scholar]
- Larger, P.J.; Breda, M.; Fraier, D.; Hughes, H.; James, C.A. Ion-suppression effects in liquid chromatography-tandem mass spectrometry due to a formulation agent, a case study in drug discovery bioanalysis. J. Pharmaceut. Biomed. Anal. 2005, 39, 206–216. [Google Scholar]
- Lin, Z.; Vicente Goncalves, C.M.; Dai, L.; Lu, H.M.; Huang, J.H.; Ji, H.; Wang, D.S.; Yi, L.Z.; Liang, Y.Z. Exploring metabolic syndrome serum profiling based on gas chromatography mass spectrometry and random forest models. Anal. Chim. Acta 2014, 827, 22–27. [Google Scholar]
- Chang, K.L.; New, L.S.; Mal, M.; Goh, C.W.; Aw, C.C.; Browne, E.R.; Chan, E.C. Metabolic profiling of 3-nitropropionic acid early-stage huntington’s disease rat model using gas chromatography time-of-flight mass spectrometry. J. Proteome Res. 2011, 10, 2079–2087. [Google Scholar]
- Halket, J.M.; Waterman, D.; Przyborowska, A.M.; Patel, R.K.; Fraser, P.D.; Bramley, P.M. Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS. J. Exp.Bot. 2005, 56, 219–243. [Google Scholar]
- Oberacher, H.; Whitley, G.; Berger, B. Evaluation of the sensitivity of the “wiley registry of tandem mass spectral data, MSforID” with MS/MS data of the “NIST/NIH/EPA mass spectral library”. J. Mass Spectrom. 2013, 48, 487–496. [Google Scholar]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. Massbank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar]
- Heinonen, M.; Shen, H.; Zamboni, N.; Rousu, J. Metabolite identification and molecular fingerprint prediction through machine learning. Bioinformatics 2012, 28, 2333–2341. [Google Scholar]
- Gravel, S.P.; Andrzejewski, S.; Avizonis, D.; St-Pierre, J. Stable isotope tracer analysis in isolated mitochondria from mammalian systems. Metabolites 2014, 4, 166–183. [Google Scholar]
- Soga, T. Capillary electrophoresis-mass spectrometry for metabolomics. Meth. Mol. Biol. 2007, 358, 129–137. [Google Scholar]
- World Health Organization (WHO). Obesity. Available online: http://www.who.int/topics/obesity/en/ (accessed on 2 September 2014).
- Wronska, A.; Kmiec, Z. Structural and biochemical characteristics of various white adipose tissue depots. Acta Physiol. 2012, 205, 194–208. [Google Scholar]
- Duncan, R.E.; Ahmadian, M.; Jaworski, K.; Sarkadi-Nagy, E.; Sul, H.S. Regulation of lipolysis in adipocytes. Ann. Rev. Nutr. 2007, 27, 79–101. [Google Scholar]
- Lehr, S.; Hartwig, S.; Lamers, D.; Famulla, S.; Muller, S.; Hanisch, F.G.; Cuvelier, C.; Ruige, J.; Eckardt, K.; Ouwens, D.M.; et al. Identification and validation of novel adipokines released from primary human adipocytes. Mol. Cell. Proteom. 2012. [Google Scholar] [CrossRef]
- Trayhurn, P.; Wood, I.S. Adipokines: Inflammation and the pleiotropic role of white adipose tissue. Br. J. Nutr. 2004, 92, 347–355. [Google Scholar]
- Molina, H.; Yang, Y.; Ruch, T.; Kim, J.W.; Mortensen, P.; Otto, T.; Nalli, A.; Tang, Q.Q.; Lane, M.D.; Chaerkady, R.; et al. Temporal profiling of the adipocyte proteome during differentiation using a five-plex silac based strategy. J. Proteome Res. 2009, 8, 48–58. [Google Scholar]
- Zhong, J.; Krawczyk, S.A.; Chaerkady, R.; Huang, H.; Goel, R.; Bader, J.S.; Wong, G.W.; Corkey, B.E.; Pandey, A. Temporal profiling of the secretome during adipogenesis in humans. J. Proteome Res. 2010, 9, 5228–5238. [Google Scholar]
- Alvarez-Llamas, G.; Szalowska, E.; de Vries, M.P.; Weening, D.; Landman, K.; Hoek, A.; Wolffenbuttel, B.H.; Roelofsen, H.; Vonk, R.J. Characterization of the human visceral adipose tissue secretome. Mol. Cell. Proteom. 2007, 6, 589–600. [Google Scholar]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar]
- Bluher, M. Adipokines—Removing road blocks to obesity and diabetes therapy. Mol. Metabol. 2014, 3, 230–240. [Google Scholar]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scime, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. Prdm16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar]
- Tang, Q.Q.; Lane, M.D. Adipogenesis: From stem cell to adipocyte. Ann. Rev. Biochem. 2012, 81, 715–736. [Google Scholar]
- Carobbio, S.; Rosen, B.; Vidal-Puig, A. Adipogenesis: New insights into brown adipose tissue differentiation. J. Mol. Endocrinol. 2013, 51, T75–T85. [Google Scholar]
- Heaton, J.M. The distribution of brown adipose tissue in the human. J. Anat. 1972, 112, 35–39. [Google Scholar]
- Saito, M.; Okamatsu-Ogura, Y.; Matsushita, M.; Watanabe, K.; Yoneshiro, T.; Nio-Kobayashi, J.; Iwanaga, T.; Miyagawa, M.; Kameya, T.; Nakada, K.; et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: Effects of cold exposure and adiposity. Diabetes 2009, 58, 1526–1531. [Google Scholar]
- Van der Lans, A.A.; Hoeks, J.; Brans, B.; Vijgen, G.H.; Visser, M.G.; Vosselman, M.J.; Hansen, J.; Jorgensen, J.A.; Wu, J.; Mottaghy, F.M.; et al. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J. Clin. Investig. 2013, 123, 3395–3403. [Google Scholar]
- Rosenwald, M.; Wolfrum, C. The origin and definition of brite versus white and classical brown adipocytes. Adipocyte 2014, 3, 4–9. [Google Scholar]
- Virtue, S.; Vidal-Puig, A. Adipose tissue expandability, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta 2013, 60, 39–43. [Google Scholar]
- Huang, P.L. A comprehensive definition for metabolic syndrome. Dis. Models Mech. 2009, 2, 231–237. [Google Scholar]
- Marchesini, G.; Moscatiello, S.; Di Domizio, S.; Forlani, G. Obesity-associated liver disease. J. Clin. Endocrinol. Metabol. 2008, 93, S74–S80. [Google Scholar]
- Lavie, C.J.; Milani, R.V.; Ventura, H.O. Obesity and cardiovascular disease: Risk factor, paradox, and impact of weight loss. J. Am. Coll. Cardiol. 2009, 53, 1925–1932. [Google Scholar]
- Ligibel, J. Obesity and breast cancer. Oncology 2011, 25, 994–1000. [Google Scholar]
- James, A.M.; Collins, Y.; Logan, A.; Murphy, M.P. Mitochondrial oxidative stress and the metabolic syndrome. Trends Endocrinol. Metabol. 2012, 23, 429–434. [Google Scholar]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar]
- Pereira, S.; Park, E.; Mori, Y.; Haber, C.A.; Han, P.; Uchida, T.; Stavar, L.; Oprescu, A.I.; Koulajian, K.; Ivovic, A.; et al. Ffa-induced hepatic insulin resistance in vivo is mediated by pkc-delta, nadph oxidase, and oxidative stress. Am. J. Physiol. Endocrinol. Metabol. 2014, 307, E34–E46. [Google Scholar]
- Hirao, K.; Maruyama, T.; Ohno, Y.; Hirose, H.; Shimada, A.; Takei, I.; Murata, M.; Morii, T.; Eguchi, T.; Hayashi, M.; et al. Association of increased reactive oxygen species production with abdominal obesity in type 2 diabetes. Obes. Res. Clin. Pract. 2010, 4, e83–e162. [Google Scholar]
- Degasperi, G.R.; Denis, R.G.; Morari, J.; Solon, C.; Geloneze, B.; Stabe, C.; Pareja, J.C.; Vercesi, A.E.; Velloso, L.A. Reactive oxygen species production is increased in the peripheral blood monocytes of obese patients. Metabolism 2009, 58, 1087–1095. [Google Scholar]
- Lefort, N.; Glancy, B.; Bowen, B.; Willis, W.T.; Bailowitz, Z.; De Filippis, E.A.; Brophy, C.; Meyer, C.; Hojlund, K.; Yi, Z.; et al. Increased reactive oxygen species production and lower abundance of complex i subunits and carnitine palmitoyltransferase 1b protein despite normal mitochondrial respiration in insulin-resistant human skeletal muscle. Diabetes 2010, 59, 2444–2452. [Google Scholar]
- Dandona, P.; Mohanty, P.; Hamouda, W.; Ghanim, H.; Aljada, A.; Garg, R.; Kumar, V. Inhibitory effect of a two day fast on reactive oxygen species (ros) generation by leucocytes and plasma ortho-tyrosine and meta-tyrosine concentrations. J. Clin. Endocrinol. Metabol. 2001, 86, 2899–2902. [Google Scholar]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar]
- Ritov, V.B.; Menshikova, E.V.; He, J.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 2005, 54, 8–14. [Google Scholar]
- Rong, J.X.; Qiu, Y.; Hansen, M.K.; Zhu, L.; Zhang, V.; Xie, M.; Okamoto, Y.; Mattie, M.D.; Higashiyama, H.; Asano, S.; et al. Adipose mitochondrial biogenesis is suppressed in db/db and high-fat diet-fed mice and improved by rosiglitazone. Diabetes 2007, 56, 1751–1760. [Google Scholar]
- Wilson-Fritch, L.; Nicoloro, S.; Chouinard, M.; Lazar, M.A.; Chui, P.C.; Leszyk, J.; Straubhaar, J.; Czech, M.P.; Corvera, S. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J. Clin. Investig. 2004, 114, 1281–1289. [Google Scholar]
- Lee, J.Y.; Lee, D.C.; Im, J.A.; Lee, J.W. Mitochondrial DNA copy number in peripheral blood is independently associated with visceral fat accumulation in healthy young adults. Int. J. Endocrinol. 2014. [Google Scholar] [CrossRef]
- Niemann, B.; Chen, Y.; Teschner, M.; Li, L.; Silber, R.E.; Rohrbach, S. Obesity induces signs of premature cardiac aging in younger patients: The role of mitochondria. J. Am. Coll. Cardiol. 2011, 57, 577–585. [Google Scholar]
- Valerio, A.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Pisconti, A.; Palomba, L.; Cantoni, O.; Clementi, E.; Moncada, S.; et al. Tnf-alpha downregulates enos expression and mitochondrial biogenesis in fat and muscle of obese rodents. J. Clin. Investig. 2006, 116, 2791–2798. [Google Scholar]
- Zorzano, A.; Hernandez-Alvarez, M.I.; Palacin, M.; Mingrone, G. Alterations in the mitochondrial regulatory pathways constituted by the nuclear co-factors pgc-1alpha or pgc-1beta and mitofusin 2 in skeletal muscle in type 2 diabetes. Biochim. Biophys. Acta 2010, 1797, 1028–1033. [Google Scholar]
- Hakansson, J.; Eliasson, B.; Smith, U.; Enerback, S. Adipocyte mitochondrial genes and the forkhead factor foxc2 are decreased in type 2 diabetes patients and normalized in response to rosiglitazone. Diabetol. Metab. Syndr. 2011, 3. [Google Scholar] [CrossRef]
- Austin, S.; St-Pierre, J. Pgc1alpha and mitochondrial metabolism—Emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar]
- Sharma, K.; Karl, B.; Mathew, A.V.; Gangoiti, J.A.; Wassel, C.L.; Saito, R.; Pu, M.; Sharma, S.; You, Y.H.; Wang, L.; et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol. 2013, 24, 1901–1912. [Google Scholar]
- Fonseca, V.; Rosenstock, J.; Patwardhan, R.; Salzman, A. Effect of metformin and rosiglitazone combination therapy in patients with type 2 diabetes mellitus: A randomized controlled trial. J. Am. Med. Assoc. 2000, 283, 1695–1702. [Google Scholar]
- Chanseaume, E.; Barquissau, V.; Salles, J.; Aucouturier, J.; Patrac, V.; Giraudet, C.; Gryson, C.; Duche, P.; Boirie, Y.; Chardigny, J.M.; et al. Muscle mitochondrial oxidative phosphorylation activity, but not content, is altered with abdominal obesity in sedentary men: Synergism with changes in insulin sensitivity. J. Clin. Endocrinol. Metabol. 2010, 95, 2948–2956. [Google Scholar]
- Shen, X.; Zheng, S.; Thongboonkerd, V.; Xu, M.; Pierce, W.M., Jr.; Klein, J.B.; Epstein, P.N. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E896–E905. [Google Scholar]
- Kim, J.A.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar]
- Petersen, P. Abnormal mitochondria in hepatocytes in human fatty liver. Acta Pathol. Microbiol. Scand. 1977, 85, 413–420. [Google Scholar]
- Eura, Y.; Ishihara, N.; Yokota, S.; Mihara, K. Two mitofusin proteins, mammalian homologues of fzo, with distinct functions are both required for mitochondrial fusion. J. Biochem. 2003, 134, 333–344. [Google Scholar]
- Bach, D.; Pich, S.; Soriano, F.X.; Vega, N.; Baumgartner, B.; Oriola, J.; Daugaard, J.R.; Lloberas, J.; Camps, M.; Zierath, J.R.; et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J. Biol. Chem. 2003, 278, 17190–17197. [Google Scholar]
- Liu, R.; Jin, P.; LiqunYu; Wang, Y.; Han, L.; Shi, T.; Li, X. Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PloS One 2014, 9, e92810. [Google Scholar]
- Hackenbrock, C.R. Chemical and physical fixation of isolated mitochondria in low-energy and high-energy states. Proc. Natl. Acad. Sci. USA 1968, 61, 598–605. [Google Scholar]
- Mogensen, M.; Sahlin, K.; Fernstrom, M.; Glintborg, D.; Vind, B.F.; Beck-Nielsen, H.; Hojlund, K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 2007, 56, 1592–1599. [Google Scholar]
- Martin, S.D.; Morrison, S.; Konstantopoulos, N.; McGee, S.L. Mitochondrial dysfunction has divergent, cell type-dependent effects on insulin action. Mol. Metab. 2014, 3, 408–418. [Google Scholar]
- Sanz, M.N.; Sanchez-Martin, C.; Detaille, D.; Vial, G.; Rigoulet, M.; El-Mir, M.Y.; Rodriguez-Villanueva, G. Acute mitochondrial actions of glitazones on the liver: A crucial parameter for their antidiabetic properties. Cell. Physiol. Biochem. 2011, 28, 899–910. [Google Scholar]
- Brunmair, B.; Staniek, K.; Gras, F.; Scharf, N.; Althaym, A.; Clara, R.; Roden, M.; Gnaiger, E.; Nohl, H.; Waldhausl, W.; et al. Thiazolidinediones, like metformin, inhibit respiratory complex i: A common mechanism contributing to their antidiabetic actions? Diabetes 2004, 53, 1052–1059. [Google Scholar]
- Pagel-Langenickel, I.; Schwartz, D.R.; Arena, R.A.; Minerbi, D.C.; Johnson, D.T.; Waclawiw, M.A.; Cannon, R.O., 3rd; Balaban, R.S.; Tripodi, D.J.; Sack, M.N. A discordance in rosiglitazone mediated insulin sensitization and skeletal muscle mitochondrial content/activity in type 2 diabetes mellitus. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2659–H2666. [Google Scholar]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metabol. 2009, 9, 311–326. [Google Scholar]
- Prentice, K.J.; Luu, L.; Allister, E.M.; Liu, Y.; Jun, L.S.; Sloop, K.W.; Hardy, A.B.; Wei, L.; Jia, W.; Fantus, I.G.; et al. The furan fatty acid metabolite cmpf is elevated in diabetes and induces beta cell dysfunction. Cell Metabol. 2014, 19, 653–666. [Google Scholar]
- Sampey, B.P.; Freemerman, A.J.; Zhang, J.; Kuan, P.F.; Galanko, J.A.; O’Connell, T.M.; Ilkayeva, O.R.; Muehlbauer, M.J.; Stevens, R.D.; Newgard, C.B.; et al. Metabolomic profiling reveals mitochondrial-derived lipid biomarkers that drive obesity-associated inflammation. PloS One 2012. [Google Scholar] [CrossRef]
- Cummins, T.D.; Holden, C.R.; Sansbury, B.E.; Gibb, A.A.; Shah, J.; Zafar, N.; Tang, Y.; Hellmann, J.; Rai, S.N.; Spite, M.; et al. Metabolic remodeling of white adipose tissue in obesity. Am. J. Physiol. Endocrinol. Metabol. 2014, 307, E262–E277. [Google Scholar]
- Won, E.Y.; Yoon, M.K.; Kim, S.W.; Jung, Y.; Bae, H.W.; Lee, D.; Park, S.G.; Lee, C.H.; Hwang, G.S.; Chi, S.W. Gender-specific metabolomic profiling of obesity in leptin-deficient ob/ob mice by 1h nmr spectroscopy. PloS One 2013. [Google Scholar] [CrossRef]
- Dudzik, D.; Zorawski, M.; Skotnicki, M.; Zarzycki, W.; Kozlowska, G.; Bibik-Malinowska, K.; Vallejo, M.; Garcia, A.; Barbas, C.; Ramos, M.P. Metabolic fingerprint of gestational diabetes mellitus. J. Proteomics 2014, 103, 57–71. [Google Scholar]
- Eisinger, K.; Krautbauer, S.; Hebel, T.; Schmitz, G.; Aslanidis, C.; Liebisch, G.; Buechler, C. Lipidomic analysis of the liver from high-fat diet induced obese mice identifies changes in multiple lipid classes. Exp. Mol. Pathol. 2014, 97, 37–43. [Google Scholar]
- Reinehr, T.; Wolters, B.; Knop, C.; Lass, N.; Hellmuth, C.; Harder, U.; Peissner, W.; Wahl, S.; Grallert, H.; Adamski, J.; et al. Changes in the serum metabolite profile in obese children with weight loss. Eur. J. Nutr. Available online: http://www.ncbi.nlm.nih.gov/pubmed/24740590 (accessed on 17 April 2014).
- Schafer, N.; Yu, Z.; Wagener, A.; Millrose, M.K.; Reissmann, M.; Bortfeldt, R.; Dieterich, C.; Adamski, J.; Wang-Sattler, R.; Illig, T.; et al. Changes in metabolite profiles caused by genetically determined obesity in mice. Metabolomics 2014, 10, 461–472. [Google Scholar]
- Li, F.; Jiang, C.; Larsen, M.C.; Bushkofsky, J.; Krausz, K.W.; Wang, T.; Jefcoate, C.R.; Gonzalez, F.J. Lipidomics reveals a link between cyp1b1 and scd1 in promoting obesity. J. Proteome Res. 2014, 13, 2679–2687. [Google Scholar]
- Suganami, T.; Tanaka, M.; Ogawa, Y. Adipose tissue inflammation and ectopic lipid accumulation. Endocr. J. 2012, 59, 849–857. [Google Scholar]
- Nobuhara, M.; Saotome, M.; Watanabe, T.; Urushida, T.; Katoh, H.; Satoh, H.; Funaki, M.; Hayashi, H. Mitochondrial dysfunction caused by saturated fatty acid loading induces myocardial insulin-resistance in differentiated h9c2 myocytes: A novel ex vivo myocardial insulin-resistance model. Exp. Cell Res. 2013, 319, 955–966. [Google Scholar]
- Yang, C.; Aye, C.C.; Li, X.; Diaz Ramos, A.; Zorzano, A.; Mora, S. Mitochondrial dysfunction in insulin resistance: Differential contributions of chronic insulin and saturated fatty acid exposure in muscle cells. Biosci. Rep. 2012, 32, 465–478. [Google Scholar]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.; Newgard, C.B.; et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metabol. 2008, 7, 45–56. [Google Scholar]
- Sevastou, I.; Kaffe, E.; Mouratis, M.A.; Aidinis, V. Lysoglycerophospholipids in chronic inflammatory disorders: The PLA(2)/LPC and ATX/LPA axes. Biochim. Biophys. Acta 2013, 1831, 42–60. [Google Scholar]
- Moolenaar, W.H.; Hla, T. Snapshot: Bioactive lysophospholipids. Cell 2012, 148, 378. [Google Scholar]
- Basanez, G.; Sharpe, J.C.; Galanis, J.; Brandt, T.B.; Hardwick, J.M.; Zimmerberg, J. Bax-type apoptotic proteins porate pure lipid bilayers through a mechanism sensitive to intrinsic monolayer curvature. J. Biol. Chem. 2002, 277, 49360–49365. [Google Scholar]
- Kalous, M.; Rauchova, H.; Drahota, Z. The effect of lysophosphatidylcholine on the activity of various mitochondrial enzymes. Biochim. Biophys. Acta 1992, 1098, 167–171. [Google Scholar]
- Kakisaka, K.; Cazanave, S.C.; Fingas, C.D.; Guicciardi, M.E.; Bronk, S.F.; Werneburg, N.W.; Mott, J.L.; Gores, G.J. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. Am. J. Physiol. 2012, 302, G77–G84. [Google Scholar]
- Eisinger, K.; Liebisch, G.; Schmitz, G.; Aslanidis, C.; Krautbauer, S.; Buechler, C. Lipidomic analysis of serum from high fat diet induced obese mice. Int. J. Mol. Sci. 2014, 15, 2991–3002. [Google Scholar]
- Nestel, P.J.; Straznicky, N.; Mellett, N.A.; Wong, G.; de Souza, D.P.; Tull, D.L.; Barlow, C.K.; Grima, M.T.; Meikle, P.J. Specific plasma lipid classes and phospholipid fatty acids indicative of dairy food consumption associate with insulin sensitivity. Am. J. Clin. Nutr. 2014, 99, 46–53. [Google Scholar]
- Watkins, S.M.; Reifsnyder, P.R.; Pan, H.J.; German, J.B.; Leiter, E.H. Lipid metabolome-wide effects of the ppargamma agonist rosiglitazone. J. Lipid Res. 2002, 43, 1809–1817. [Google Scholar]
- Bao, Y.; Zhao, T.; Wang, X.; Qiu, Y.; Su, M.; Jia, W. Metabonomic variations in the drug-treated type 2 diabetes mellitus patients and healthy volunteers. J. Proteome Res. 2009, 8, 1623–1630. [Google Scholar]
- Zhu, Y.; Feng, Y.; Shen, L.; Xu, D.; Wang, B.; Ruan, K.; Cong, W. Effect of metformin on the urinary metabolites of diet-induced-obese mice studied by ultra performance liquid chromatography coupled to time-of-flight mass spectrometry (UPLC-TOF/MS). J. Chromatogr. B 2013, 925, 110–116. [Google Scholar]
- Huo, T.; Cai, S.; Lu, X.; Sha, Y.; Yu, M.; Li, F. Metabonomic study of biochemical changes in the serum of type 2 diabetes mellitus patients after the treatment of metformin hydrochloride. J. Pharmaceut. Biomed. Anal. 2009, 49, 976–982. [Google Scholar]
- Simon-Szabo, L.; Kokas, M.; Mandl, J.; Keri, G.; Csala, M. Metformin attenuates palmitate-induced endoplasmic reticulum stress, serine phosphorylation of irs-1 and apoptosis in rat insulinoma cells. PloS One 2014. [Google Scholar] [CrossRef]
- Clarke, C.; Xiao, R.; Place, E.; Zhang, Z.; Sondheimer, N.; Bennett, M.; Yudkoff, M.; Falk, M.J. Mitochondrial respiratory chain disease discrimination by retrospective cohort analysis of blood metabolites. Mol. Genet. Metab. 2013, 110, 145–152. [Google Scholar]
- Eaton, S. Control of mitochondrial beta-oxidation flux. Prog. Lipid Res. 2002, 41, 197–239. [Google Scholar]
- Shah, S.H.; Crosslin, D.R.; Haynes, C.S.; Nelson, S.; Turer, C.B.; Stevens, R.D.; Muehlbauer, M.J.; Wenner, B.R.; Bain, J.R.; Laferrere, B.; et al. Branched-chain amino acid levels are associated with improvement in insulin resistance with weight loss. Diabetologia 2012, 55, 321–330. [Google Scholar]
- She, P.; Reid, T.M.; Bronson, S.K.; Vary, T.C.; Hajnal, A.; Lynch, C.J.; Hutson, S.M. Disruption of bcatm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cell Metab. 2007, 6, 181–194. [Google Scholar]
- Walford, G.A.; Davis, J.; Warner, A.S.; Ackerman, R.J.; Billings, L.K.; Chamarthi, B.; Fanelli, R.R.; Hernandez, A.M.; Huang, C.; Khan, S.Q.; et al. Branched chain and aromatic amino acids change acutely following two medical therapies for type 2 diabetes mellitus. Metabolism 2013, 62, 1772–1778. [Google Scholar]
- Hsiao, G.; Chapman, J.; Ofrecio, J.M.; Wilkes, J.; Resnik, J.L.; Thapar, D.; Subramaniam, S.; Sears, D.D. Multi-tissue, selective ppargamma modulation of insulin sensitivity and metabolic pathways in obese rats. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E164–E174. [Google Scholar]
- Yang, R.; Dong, J.; Zhao, H.; Li, H.; Guo, H.; Wang, S.; Zhang, C.; Wang, S.; Wang, M.; Yu, S.; et al. Association of branched-chain amino acids with carotid intima-media thickness and coronary artery disease risk factors. PloS One 2014. [Google Scholar] [CrossRef]
- Seifert, E.L.; Fiehn, O.; Bezaire, V.; Bickel, D.R.; Wohlgemuth, G.; Adams, S.H.; Harper, M.E. Long-chain fatty acid combustion rate is associated with unique metabolite profiles in skeletal muscle mitochondria. PloS One 2010. [Google Scholar] [CrossRef]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2010, 33, S62–S69. [Google Scholar]
- Gohring, I.; Sharoyko, V.V.; Malmgren, S.; Andersson, L.E.; Spegel, P.; Nicholls, D.G.; Mulder, H. Chronic high glucose and pyruvate levels differentially affect mitochondrial bioenergetics and fuel-stimulated insulin secretion from clonal ins-1 832/13 cells. J. Biol. Chem. 2014, 289, 3786–3798. [Google Scholar]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and ntps in cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10574–10579. [Google Scholar]
- Jang, E.H.; Kim, H.K.; Park, C.S.; Kang, J.H. Increased expression of hepatic organic cation transporter 1 and hepatic distribution of metformin in high-fat diet-induced obese mice. Drug Metabol. Pharmacokinet. 2010, 25, 392–397. [Google Scholar]
- Chen, L.; Shu, Y.; Liang, X.; Chen, E.C.; Yee, S.W.; Zur, A.A.; Li, S.; Xu, L.; Keshari, K.R.; Lin, M.J.; et al. Oct1 is a high-capacity thiamine transporter that regulates hepatic steatosis and is a target of metformin. Proc. Natl. Acad. Sci. USA 2014, 111, 9983–9988. [Google Scholar]
- Abdul-Ghani, M.A.; Muller, F.L.; Liu, Y.; Chavez, A.O.; Balas, B.; Zuo, P.; Chang, Z.; Tripathy, D.; Jani, R.; Molina-Carrion, M.; et al. Deleterious action of fa metabolites on atp synthesis: Possible link between lipotoxicity, mitochondrial dysfunction, and insulin resistance. Am. J. Physiol. Endocrinol. Metabol. 2008, 295, E678–E685. [Google Scholar]
- Zheng, H.; Yde, C.C.; Arnberg, K.; Molgaard, C.; Michaelsen, K.F.; Larnkjaer, A.; Bertram, H.C. Nmr-based metabolomic profiling of overweight adolescents: An elucidation of the effects of inter-/intraindividual differences, gender, and pubertal development. BioMed Research Int. 2014, 2014. Article 537157. [Google Scholar]
- Gkourogianni, A.; Kosteria, I.; Telonis, A.G.; Margeli, A.; Mantzou, E.; Konsta, M.; Loutradis, D.; Mastorakos, G.; Papassotiriou, I.; Klapa, M.I.; et al. Plasma metabolomic profiling suggests early indications for predisposition to latent insulin resistance in children conceived by ICSI. PloS One 2014. [Google Scholar] [CrossRef]
- Ekberg, N.R.; Brismar, K.; Malmstedt, J.; Hedblad, M.A.; Adamson, U.; Ungerstedt, U.; Wisniewski, N. Analyte flux at a biomaterial-tissue interface over time: Implications for sensors for type 1 and 2 diabetes mellitus. J. Diabetes Sci. Technol. 2010, 4, 1063–1072. [Google Scholar]
- Sorriento, D.; Pascale, A.V.; Finelli, R.; Carillo, A.L.; Annunziata, R.; Trimarco, B.; Iaccarino, G. Targeting mitochondria as therapeutic strategy for metabolic disorders. Sci. World J. 2014, 2014. Article 604685. [Google Scholar]
- Jones, L.R.; Wilson, C.I.; Wadden, T.A. Lifestyle modification in the treatment of obesity: An educational challenge and opportunity. Clin. Pharmacol. Therapeut. 2007, 81, 776–779. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Demine, S.; Reddy, N.; Renard, P.; Raes, M.; Arnould, T. Unraveling Biochemical Pathways Affected by Mitochondrial Dysfunctions Using Metabolomic Approaches. Metabolites 2014, 4, 831-878. https://doi.org/10.3390/metabo4030831
Demine S, Reddy N, Renard P, Raes M, Arnould T. Unraveling Biochemical Pathways Affected by Mitochondrial Dysfunctions Using Metabolomic Approaches. Metabolites. 2014; 4(3):831-878. https://doi.org/10.3390/metabo4030831
Chicago/Turabian StyleDemine, Stéphane, Nagabushana Reddy, Patricia Renard, Martine Raes, and Thierry Arnould. 2014. "Unraveling Biochemical Pathways Affected by Mitochondrial Dysfunctions Using Metabolomic Approaches" Metabolites 4, no. 3: 831-878. https://doi.org/10.3390/metabo4030831
APA StyleDemine, S., Reddy, N., Renard, P., Raes, M., & Arnould, T. (2014). Unraveling Biochemical Pathways Affected by Mitochondrial Dysfunctions Using Metabolomic Approaches. Metabolites, 4(3), 831-878. https://doi.org/10.3390/metabo4030831