The Effect of LC-MS Data Preprocessing Methods on the Selection of Plasma Biomarkers in Fed vs. Fasted Rats

Abstract
:1. Introduction
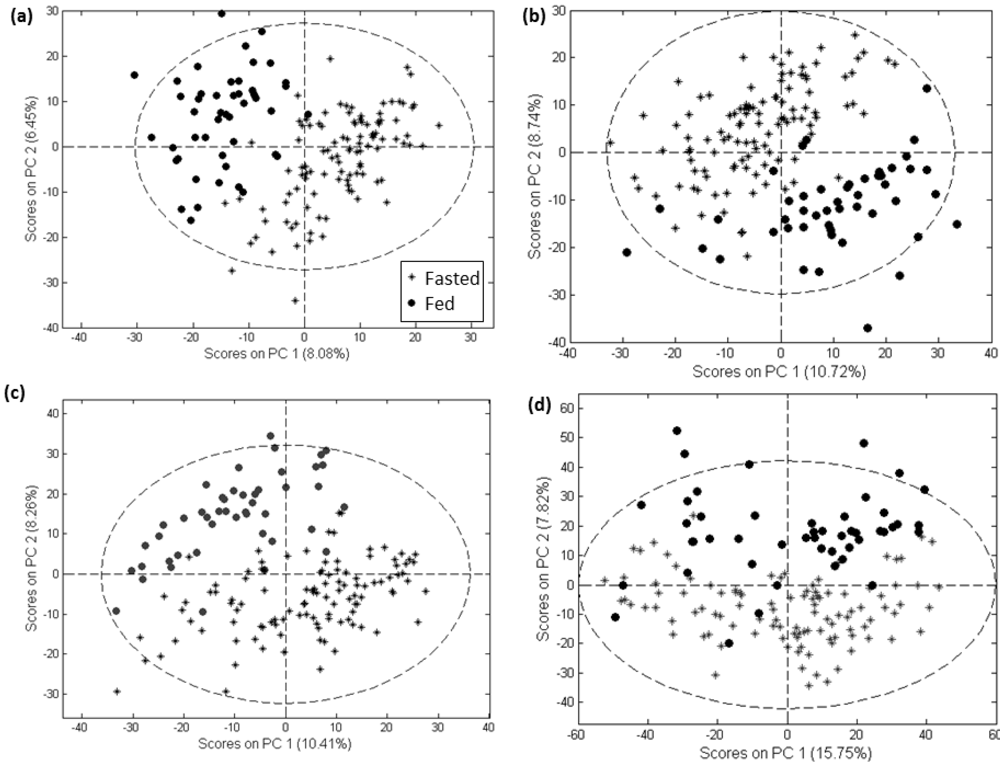
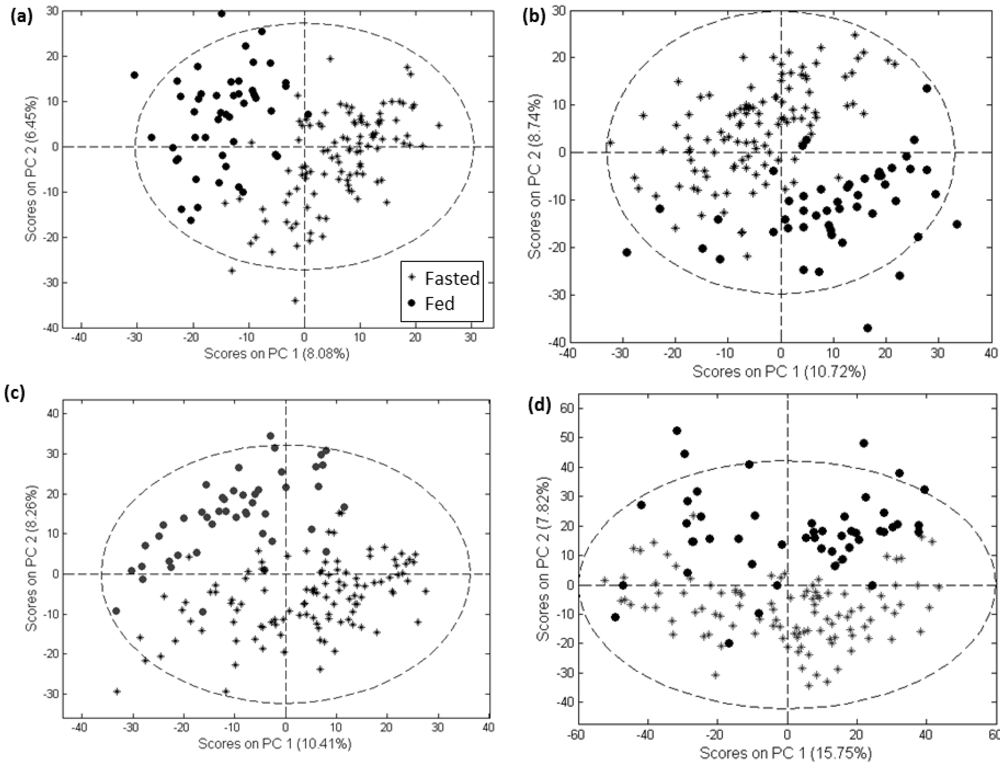
2. Results and Discussion
2.1. Comparison of Data Preprocessing Methods
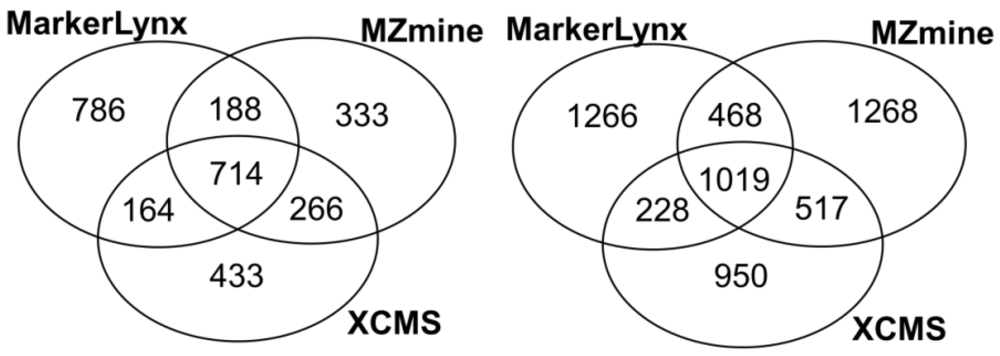

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO | RT (min) | Measured m/z | MX Rank | MZ Rank | XCMS Rank | Custom rank | Group | Suggested Compound | Adduct | Monoisotopic mass |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.64 | 105.02 | 57 | 17 | 14 | 194 | fed | U1 | ||
| 2 | 0.82 | 116.07 | 91 | 26 | 17 | 507 | fed | U2 | ||
| 3 | 1.15 | 180.06 | 67 | 28 | 21 | 27 | fed | U3 | ||
| 4 | 1.15 | 383.12 | 40 | 80 | 25 | 624 | fed | U3 | ||
| 5 | 1.36 | 59.01 | 21 | 34 | 9 | 7 | fasted | 3-hydroxybutanoic acid F | 104.0473 | |
| 6 | 1.36 | 260.00 | 49 | 68 | nd | 22 | fasted | 3-hydroxybutanoic acid F | 104.0473 | |
| 7 | 1.37 | 229.07 | 20 | 35 | nd | 72 | fasted | 3-hydroxybutanoic acid A | [2M+Na-H] | 104.0473 |
| 8 | 1.37 | 103.04 | 39 | 15 | nd | 20 | fasted | 3-hydroxybutanoic acid | [M-H] | 104.0473 |
| 9 | 1.37 | 261.18 | 1424 | nd | 18 | 14 | fed | Isoleucine | [2M-H] | 131.0946 |
| 10 | 1.37 | 130.09 | 25 | nd | 24 | 65 | fed | Isoleucine | [M-H]- | 131.0946 |
| 11 | 1.80 | 178.05 | nd | 22 | nd | 166 | fed | U4 | ||
| 12 | 1.88 | 134.06 | 14 | 9 | 6 | 40 | fasted | Hippuric acid * F | 179.0582 | |
| 13 | 1.88 | 178.05 | 15 | 7 | 4 | 116 | fasted | Hippuric acid * | [M-H] | 179.0582 |
| 14 | 2.02 | 344.10 | 383 | nd | 222 | 12 | none | U5 | ||
| 15 | 2.46 | 365.07 | 3 | 6 | nd | 43 | fed | U6 | ||
| 16 | 2.46 | 623.36 | 8 | nd | 3 | 94 | fed | U6 | ||
| 17 | 2.46 | 343.08 | 2 | 2 | 1 | 6 | fed | U6 | ||
| 18 | 2.47 | 623.87 | 4 | nd | nd | 16 | fed | U7 | ||
| 19 | 3.00 | 185.12 | 793 | 23 | 77 | 284 | fed | U8 | ||
| 20 | 3.50 | 505.30 | 1833 | nd | nd | 10 | none | U9 | ||
| 21 | 4.11 | 586.31 | nd | 13 | nd | 13 | fed | LPC(20:5) | [M+FA-H] | 541.3168 |
| 22 | 4.12 | 309.20 | 1 | 10 | 7 | 1802 | fed | LPC(20:5) F | 541.3168 | |
| 23 | 4.15 | 452.28 | 22 | 30 | 22 | 1006 | fed | LPC(14:0) F | 467.3012 | |
| 24 | 4.16 | 512.30 | 17 | 21 | 19 | 45 | fed | LPC(14:0) A | [M+FA-H] | 467.3012 |
| 25 | 4.16 | 979.60 | 19 | nd | nd | 33 | fed | LPC(14:0) A | [2M+FA-H] | 467.3012 |
| 26 | 4.17 | 502.29 | 13 | 11 | nd | 25 | fed | LPC(18:3) F | 517.3168 | |
| 27 | 4.18 | 562.31 | 5 | 8 | 51 | 17 | fed | LPC(18:3) | [M+FA-H] | 517.3168 |
| 28 | 4.18 | 818.50 | 16 | nd | nd | 1672 | fed | U10 | ||
| 29 | 4.18 | 526.30 | 11 | 19 | 11 | 912 | fed | LPC(20:5) F | 541.3168 | |
| 30 | 4.19 | 586.31 | 7 | 18 | 8 | 13 | fed | LPC(20:5) | [M+FA-H] | 541.3168 |
| 31 | 4.23 | 563.32 | nd | nd | 13 | 15 | fed | U11 | ||
| 32 | 4.34 | 476.28 | 23 | 1 | nd | 1 | fed | 2-acyl LPC(18:2) F | 519.3325 | |
| 33 | 4.35 | 564.33 | 10 | 12 | nd | 3 | fed | 2-acyl LPC(18:2) | [M+FA-H] | 519.3325 |
| 34 | 4.35 | 504.31 | 147 | 3 | nd | 2 | fed | 2-acyl LPC(18:2) F | 519.3325 | |
| 35 | 4.35 | 578.30 | nd | 5 | nd | 35 | fasted | U12 | ||
| 36 | 4.36 | 632.33 | 120 | 25 | nd | 113 | fed | U13 | ||
| 37 | 4.38 | 281.25 | 33 | nd | 15 | nd | fasted | U14 | ||
| 38 | 4.43 | 476.28 | 105 | 4 | 2 | 1 | fed | 1-acyl LPC(18:2) F | 519.3325 | |
| 39 | 4.44 | 168.35 | 6 | nd | nd | 1512 | fed | 1-acyl LPC(18:2) F | 519.3325 | |
| 40 | 4.44 | 995.59 | 60 | nd | nd | 4 | fed | 1-acyl LPC(18:2) F | 519.3325 | |
| 41 | 4.44 | 168.63 | 18 | nd | nd | 170 | fed | 1-acyl LPC(18:2) F | 519.3325 | |
| 42 | 4.44 | 504.31 | 65 | 14 | 32 | 2 | fed | 1-acyl LPC(18:2) F | 519.3325 | |
| 43 | 4.45 | 457.10 | 12 | nd | 561 | 2332 | fasted | U15 | ||
| 44 | 4.45 | 564.33 | 32 | 31 | 20 | 3 | fed | 1-acyl LPC(18:2) | [M+FA-H] | 519.3325 |
| 45 | 4.45 | 335.40 | nd | nd | nd | 8 | none | none | ||
| 46 | 4.45 | 335.70 | nd | nd | nd | 9 | none | none | ||
| 47 | 4.45 | 477.28 | nd | nd | nd | 21 | fed | 1-acyl LPC(18:2) iso1 | ||
| 48 | 4.45 | 564.10 | nd | nd | nd | 23 | none | none | ||
| 49 | 4.45 | 565.34 | nd | nd | nd | 5 | fed | 1-acyl LPC(18:2) iso2 | ||
| 50 | 4.45 | 587.30 | nd | nd | nd | 11 | none | none | ||
| 51 | 4.45 | 996.59 | nd | nd | nd | 19 | fed | 1-acyl LPC(18:2) iso3 | ||
| 52 | 4.50 | 552.33 | 24 | 46 | 63 | 320 | fed | U16 | ||
| 53 | 4.62 | 452.28 | 48 | 55 | 23 | 1006 | fasted | U17 | ||
| 54 | 4.65 | 566.35 | 374 | 24 | nd | 138 | fed | 1-acyl LPC(18:1) | [M+FA-H] | 521.3481 |
| 55 | 4.73 | 478.29 | 9 | 16 | 12 | 18 | fed | LPE(18:1) * | [M-H] | 479.3012 |
| 56 | 4.88 | 445.33 | 76 | 20 | 10 | 1206 | fasted | U19 | ||
| 57 | 5.14 | 277.22 | 85 | 106 | 5 | 98 | fasted | Gamma-Linolenic acid * | [M-H] | 278.2246 |
| 58 | 5.22 | 338.30 | 100 | nd | nd | 24 | none | U20 | ||
| 59 | 5.38 | 279.23 | 145 | nd | 16 | 177 | fasted | Linoleic acid * | [M-H] | 280.2402 |
| NO | RT (min) | Measured m/z | MX Rank | MZ Rank | XCMS Rank | Custom rank | Group | SuggestedCompound | Suggested Adduct | Monoisotopic mass |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.53 | 112.11 | nd | 12 | 13 | 301 | fasted | U1 | ||
| 2 | 0.57 | 730.70 | 276 | nd | nd | 25 | fasted | U2 | ||
| 3 | 0.61 | 103.04 | 46 | nd | 19 | 2901 | fed | L-Carnitine *F | 161.1052 | |
| 4 | 0.61 | 102.09 | 1368 | nd | 21 | 481 | fed | L-Carnitine *F | 161.1052 | |
| 5 | 0.61 | 162.11 | 31 | 41 | 10 | 10 | fed | L-Carnitine * | [M+H] | 161.1052 |
| 6 | 0.66 | 70.07 | 12 | 11 | 25 | 22 | fed | D-proline *F | 115.0633 | |
| 7 | 0.66 | 116.07 | 13 | 14 | 12 | 11 | fed | D-proline * | [M+H] | 115.0633 |
| 8 | 0.86 | 130.09 | 24 | 521 | 44 | 838 | fasted | U3 | ||
| 9 | 0.90 | 144.10 | 23 | nd | 16 | 455 | fasted | L-Acetylcarnitine*F | 203.1158 | |
| 10 | 0.90 | 204.12 | 28 | 18 | 6 | 8 | fasted | L-Acetylcarnitine* | [M+H] | 203.1158 |
| 11 | 0.90 | 145.05 | 21 | 13 | 11 | 41 | fasted | L-Acetylcarnitine*F | 203.1158 | |
| 12 | 1.17 | 248.15 | 49 | 23 | 7 | 38 | fasted | U4 | ||
| 13 | 1.64 | 231.12 | nd | 100 | 1 | 649 | fasted | U5 | ||
| 14 | 1.90 | 105.03 | 1 | 17 | 2 | 78 | fasted | Hippuric Acid*F | 179.0582 | |
| 15 | 1.90 | 77.04 | 3 | 19 | 3 | 578 | fasted | Hippuric Acid*F | 179.0582 | |
| 16 | 2.23 | 316.21 | 19 | 46 | nd | 179 | fasted | U6 | ||
| 17 | 2.42 | 899.43 | nd | nd | nd | 17 | fed | U7 | ||
| 18 | 2.42 | 287.20 | nd | nd | nd | 1 | fed | U7 | ||
| 19 | 2.42 | 286.20 | 7 | 3 | 50 | 4 | fed | U7 | ||
| 20 | 3.42 | 536.34 | 35 | nd | nd | 24 | fed | U8 | ||
| 21 | 3.49 | 158.16 | 338 | 222 | 63 | 19 | fasted | U9 | ||
| 22 | 4.11 | 542.33 | 16 | 16 | nd | 21 | fed | LPC(20:5) | [M+H] | 541.3168 |
| 23 | 4.12 | 564.31 | nd | 15 | nd | 43 | fed | LPC(20:5) A | [M+Na] | 541.3168 |
| 24 | 4.16 | 312.03 | 151 | nd | 17 | 2659 | fed | U10 | ||
| 25 | 4.16 | 468.31 | 20 | 24 | 23 | 15 | fed | LPC(14:0) | [M+H] | 467.3012 |
| 26 | 4.19 | 540.31 | 25 | 64 | nd | 47 | fed | LPC(18:3) A | [M+Na] | 517.3168 |
| 27 | 4.19 | 518.33 | 15 | 6 | 81 | 62 | fed | LPC(18:3) | [M+H] | 517.3168 |
| 28 | 4.23 | 445.40 | nd | nd | nd | 12 | fasted | octadecanoylcarnitineIso | ||
| 29 | 4.23 | 444.37 | 18 | 33 | 47 | 33 | fasted | octadecanoylcarnitine | ||
| 30 | 4.35 | 337.28 | 9 | 9 | 5 | 57 | fed | 2-acyl LPC(18:2) F | 519.3325 | |
| 31 | 4.35 | 520.34 | 6 | 1 | nd | 2 | fed | 2-acyl LPC(18:2) | [M+H] | 519.3325 |
| 32 | 4.36 | 542.33 | 4 | 2 | nd | 21 | fed | 2-acyl LPC (18:2) A | [M+Na] | 519.3325 |
| 33 | 4.36 | 819.96 | 22 | nd | nd | 950 | fed | U11 | ||
| 34 | 4.36 | 502.33 | nd | 10 | nd | 28 | fed | 2-acyl LPC(18:2) F | [M+Na] | 479.3376 |
| 35 | 4.42 | 566.32 | 1024 | 2058 | 15 | 50 | fasted | U12 | ||
| 36 | 4.42 | 844.47 | 219 | 233 | 20 | 1312 | fasted | U13 | ||
| 37 | 4.44 | 519.90 | nd | nd | nd | 18 | fed | U14 | ||
| 38 | 4.44 | 521.35 | nd | nd | nd | 5 | fed | 1-acyl LPC(18:2) Iso1 | [M+H] | 519.3325 |
| 39 | 4.45 | 523.35 | nd | 7 | nd | 89 | fed | 1-acyl LPC(18:2)Iso2 | [M+H] | 519.3325 |
| 40 | 4.45 | 519.70 | 316 | nd | nd | 7 | fed | U15 | ||
| 41 | 4.45 | 997.64 | 14 | 20 | 9 | 3 | fed | 1-acyl LPC(18:2) A | 519.3325 | |
| 42 | 4.45 | 819.97 | 2 | 21 | 835 | 950 | fasted | U16 | ||
| 43 | 4.45 | 520.34 | 8 | 4 | 18 | 2 | fed | 1-acyl LPC(18:2) | [M+H] | 519.3325 |
| 44 | 4.45 | 998.64 | 30 | nd | nd | 6 | fed | U17 | ||
| 45 | 4.45 | 460.29 | 59 | 54 | 14 | 612 | fed | 1-acyl LPC(18:2) F | 519.3325 | |
| 46 | 4.45 | 520.10 | nd | nd | nd | 13 | none | U18 | ||
| 47 | 4.45 | 520.90 | nd | nd | nd | 23 | none | U18 | ||
| 48 | 4.45 | 521.55 | nd | nd | nd | 20 | none | U18 | ||
| 49 | 4.45 | 521.80 | nd | nd | nd | 16 | none | U18 | ||
| 50 | 4.45 | 807.97 | 5 | 8 | 4 | 2664 | fed | U19 | ||
| 51 | 4.63 | 949.64 | 34 | 25 | 48 | 85 | fasted | U20 | ||
| 52 | 4.64 | 454.30 | 32 | 22 | 22 | 1425 | fasted | U20 | ||
| 53 | 4.65 | 975.70 | 76 | nd | nd | 14 | fed | U21 | ||
| 54 | 4.65 | 522.36 | 10 | nd | nd | 70 | fed | 2-acyl LPC(18:1) * | [M+H] | 521.3481 |
| 55 | 4.65 | 339.29 | 17 | 5 | 8 | 573 | fed | 2-acyl LPC(18:1) *F | ||
| 56 | 4.68 | 520.34 | 11 | nd | 24 | 2 | fed | U22 | [M+H] | 519.3325 |
2.2. Custom Method vs. Software Tools

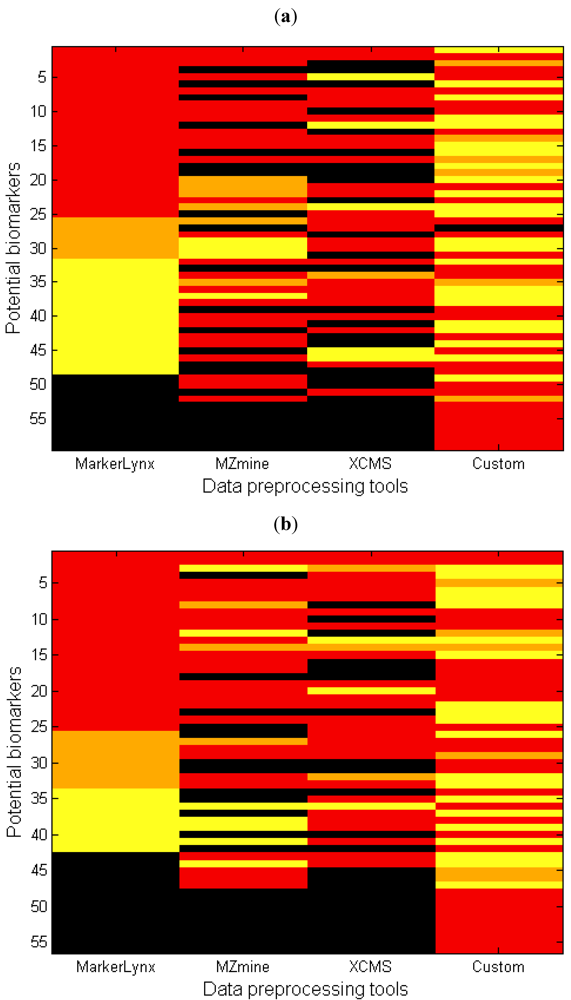
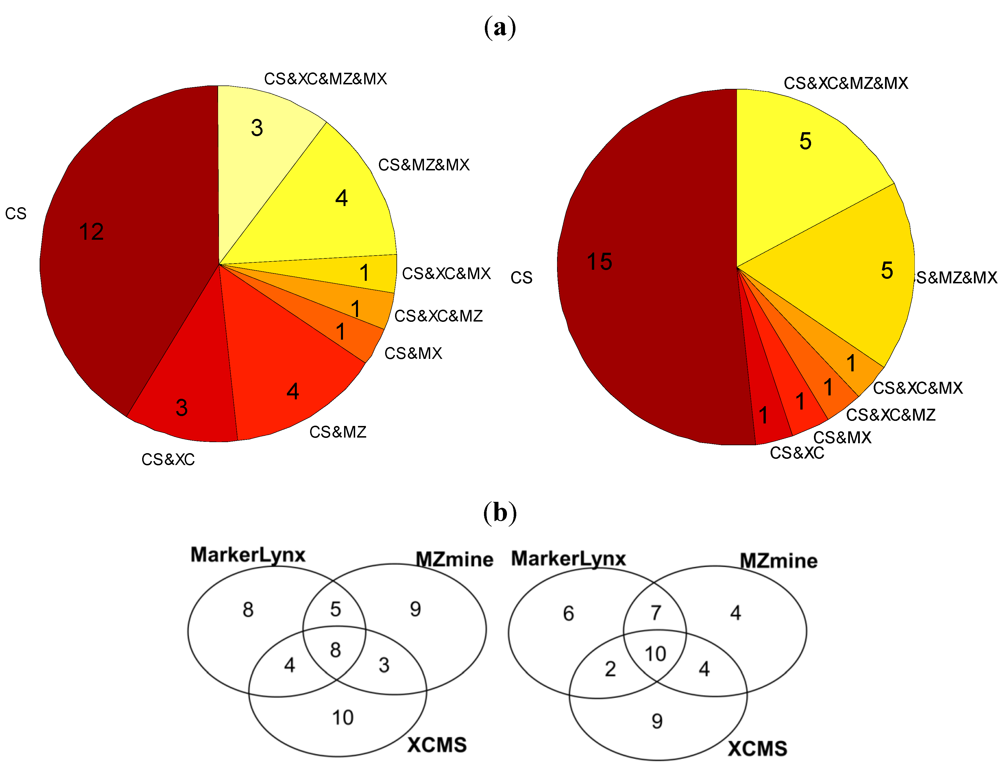
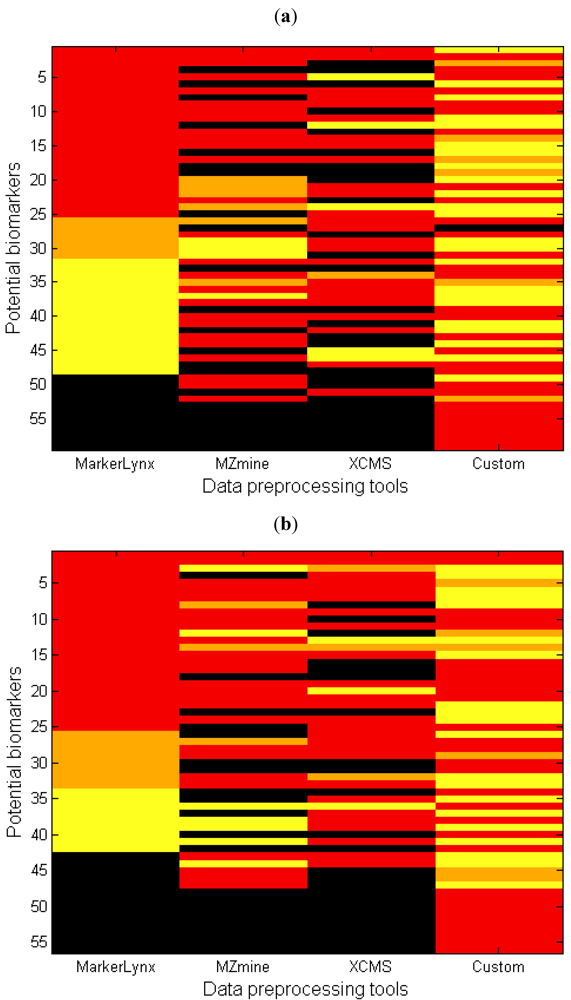
2.3. Comparison of the Dedicated Software Tools
- (1)
- (2)
- The marker is detected but the peak height assignment was not the same among software tools, which did not result in significant difference between fasted and fed states. One reason of this is shown in the next section as influence of gap filling. This condition is illustrated as yellow in Figure 4.
- (3)
- The data analysis method affected the marker selection. This was discussed as an effect of autoscaling previously. This condition is illustrated by orange in Figure 4.
2.4. The Influence of Gap Filling
2.5. Software Preprocessing Settings
2.6. Biomarker Patterns
2.7. Biomarkers of Fasted and Fed State
3. Experimental Section
3.1. Animal Study and Sample Collection
3.2. Plasma Preprocessing and LC-QTOF Analysis
3.3. Authentic Standards
3.4. Raw Data
3.5. Software Tools for Data Preprocessing
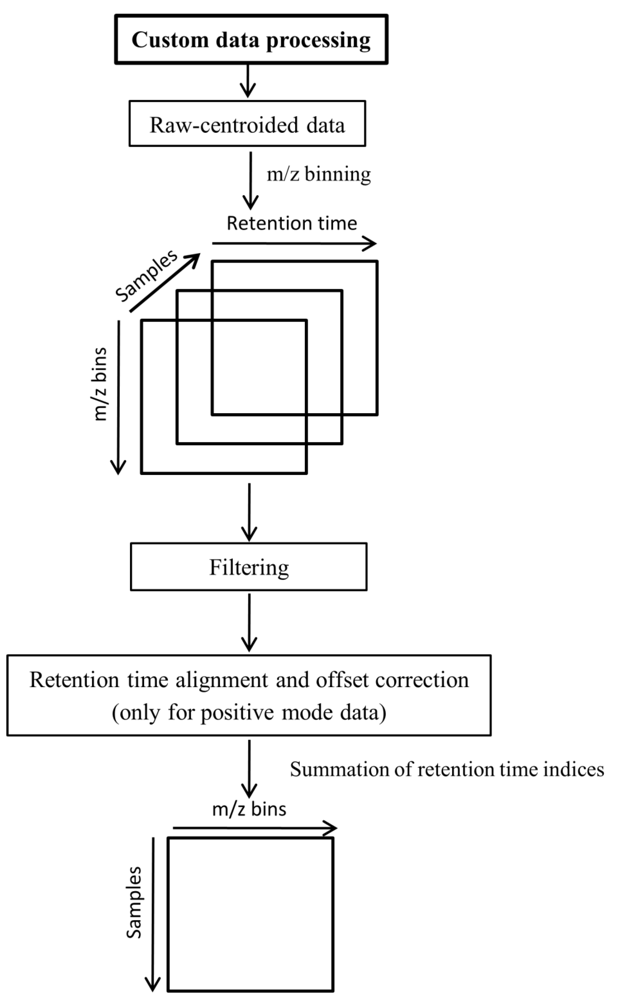
3.6. Custom Methods for Data Preprocessing
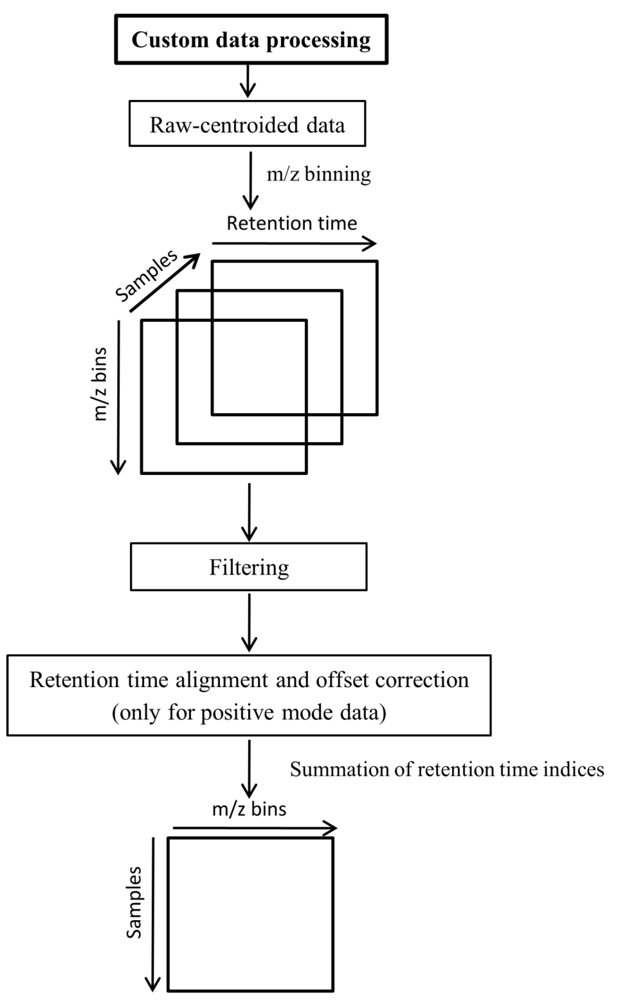
3.7. Data Analysis
3.7.1. Variable Reduction
3.7.2. Variable (Feature) Selection
3.8. Marker Identification
4. Conclusions
Supplementary Materials
Acknowledgements
References
- Zivkovic, A.M.; Wiest, M.M.; Nguyen, U.; Nording, M.L.; Watkins, S.M.; German, J.B. Assessing individual metabolic responsiveness to a lipid challenge using a targeted metabolomic approach. Metabolomics 2009, 5, 209–218. [Google Scholar] [CrossRef]
- Sharman, M.J.; Gomez, A.L.; Kraemer, W.J.; Volek, J.S. Very low-carbohydrate and low-fat diets affect fasting lipids and postprandial lipernia differently in overweight men. J. Nutr. 2004, 134, 880–885. [Google Scholar]
- Lindon, J.C.; Nicholson, J.K.; Holmes, E. The Handbook of Metabonomics and Metabolomics; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Brindle, J.T.; Nicholson, J.K.; Schofield, P.M.; Grainger, D.J.; Holmes, E. Application of chemometrics to H-1 NMR spectroscopic data to investigate a relationship between human serum metabolic profiles and hypertension. Analyst 2003, 128, 32–36. [Google Scholar]
- Constantinou, M.A.; Tsantili-Kakoulidou, A.; Andreadou, I.; Iliodromitis, E.K.; Kremastinos, D.T.; Mikros, E. Application of NMR-based metabonomics in the investigation of myocardial ischemia-reperfusion, ischemic preconditioning and antioxidant intervention in rabbits. Eur. J. Pharm. Sci. 2007, 30, 303–314. [Google Scholar] [CrossRef]
- Fardet, A.; Llorach, R.; Martin, J. F.; Besson, C.; Lyan, B.; Pujos-Guillot, E.; Scalbert, A. A liquid chromatography-quadrupole time-of-flight (LC-QTOF)-based metabolomic approach reveals new metabolic effects of catechin in rats fed high-fat diets. J. Proteome Res. 2008, 7, 2388–2398. [Google Scholar]
- Kim, J.Y.; Park, J.Y.; Kim, O.Y.; Ham, B.M.; Kim, H.J.; Kwon, D.Y.; Jang, Y.; Lee, J.H. Metabolic profiling of plasma in overweight/obese and lean men using ultra performance liquid chromatography and Q-TOF mass spectrometry (UPLC-Q-TOF MS). J. Proteome Res. 2010, 9, 4368–4375. [Google Scholar]
- Wilson, I.D.; Nicholson, J.K.; Castro-Perez, J.; Granger, J.H.; Johnson, K.A.; Smith, B.W.; Plumb, R.S. High resolution "Ultra performance" liquid chromatography coupled to oa-TOF mass spectrometry as a tool for differential metabolic pathway profiling in functional genomic studies. J. Proteome Res. 2005, 4, 591–598. [Google Scholar] [CrossRef]
- Katajamaa, M.; Oresic, M. Data processing for mass spectrometry-based metabolomics. J. Chromatogr. A 1158, 318–328. [Google Scholar]
- Castillo, S.; Gopalacharyulu, P.; Yetukuri, L.; Oresic, M. Algorithms and tools for the preprocessing of LC-MS metabolomics data. Chemom. Intell. Lab. Syst. 2011, 108, 23–32. [Google Scholar] [CrossRef]
- Yu, T.W.; Park, Y.; Johnson, J.M.; Jones, D.P. apLCMS-adaptive processing of high-resolution LC/MS data. Bioinformatics 2009, 25, 1930–1936. [Google Scholar]
- Schulz-Trieglaff, O.; Hussong, R.; Gropl, C.; Leinenbach, A.; Hildebrandt, A.; Huber, C.; Reinert, K. Computational quantification of peptides from LC-MS data. J. Comput. Biol. 2008, 15, 685–704. [Google Scholar]
- Lange, E.; Tautenhahn, R.; Neumann, S.; Gropl, C. Critical assessment of alignment procedures for LC- MS proteomics and metabolomics measurements. BMC Bioinformatics 2008, 9, 375. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 2010, 11, 395. [Google Scholar]
- Smith, C.A.; Want, E.J.; O'Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar]
- Tautenhahn, R.; Bottcher, C.; Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinformatics 2008, 9, 504. [Google Scholar]
- Nielsen, N.J.; Tomasi, G.; Frandsen, R.J.N.; Kristensen, M.B.; Nielsen, J.; Giese, H.; Christensen, J.H. A pre-processing strategy for liquid chromatography time-of-flight mass spectrometry metabolic fingerprinting data. Metabolomics 2010, 6, 341–352. [Google Scholar] [CrossRef]
- Westerhuis, J.A.; Hoefsloot, H.C.J.; Smit, S.; Vis, D.J.; Smilde, A.K.; van Velzen, E.J.J.; van Duijnhoven, J.P.M.; van Dorsten, F.A. Assessment of PLSDA cross validation. Metabolomics 2008, 4, 81–89. [Google Scholar] [CrossRef]
- Kristensen, M.; Engelsen, S.B.; Dragsted, L.O. LC-MS metabolomics top-down approach reveals new exposure and effect biomarkers of apple and apple-pectin intake. Metabolomics 2011, in press. [Google Scholar]
- Subbaiah, P.V.; Liu, M. Comparative studies on the substrate specificity of lecithin:cholesterol acyltransferase towards the molecular species of phosphatidylcholine in the plasma of 14 vertebrates. J. Lipid Res. 1996, 37, 113–122. [Google Scholar]
- Weltzien, H.U. Cytolytic and Membrane-Perturbing Properties of Lysophosphatidylcholine. Biochim. Biophys. Acta 1979, 559, 259–287. [Google Scholar] [CrossRef]
- Han, M.S.; Lim, Y.M.; Quan, W.; Kim, J.R.; Chung, K.W.; Kang, M.; Kim, S.; Park, S.Y.; Han, J.S.; Park, S.Y.; et al. Lysophosphatidylcholine as an effector of fatty acid-induced insulin resistance. J. Lipid Res. 2011, 52, 1234–1246. [Google Scholar] [CrossRef]
- Sekas, G.; Patton, G.M.; Lincoln, E.C.; Robins, S.J. Origin of plasma lysophosphatidylcholine: Evidence for direct hepatic secretion in the rat. J. Lab. Clin. Med. 1985, 105, 190–194. [Google Scholar]
- Croset, M.; Brossard, N.; Polette, A.; Lagarde, M. Characterization of plasma unsaturated lysophosphatidylcholines in human and rat. Biochem. J. 2000, 345, 61–67. [Google Scholar] [CrossRef]
- Seppanen-Laakso, T.; Oresic, M. How to study lipidomes. J. Mol. Endocrinol. 2009, 42, 185–190. [Google Scholar] [CrossRef]
- Sandra, K.; Pereira, A.D.; Vanhoenacker, G.; David, F.; Sandra, P. Comprehensive blood plasma lipidomics by liquid chromatography/quadrupole time-of-flight mass spectrometry. J. Chromatogr. A 2010, 1217, 4087–4099. [Google Scholar]
- Kerner, J.; Hoppel, C. Fatty acid import into mitochondria. Biochimica et Biophysica Acta-Mol. Cell Biol. Lipids 2000, 1486, 1–17. [Google Scholar] [CrossRef]
- Pearson, D.J.; Tubbs, P.K. Carnitine and Derivatives in Rat Tissues. Biochem. J. 1967, 105, 953–963. [Google Scholar]
- Shaham, O.; Wei, R.; Wang, T.J.; Ricciardi, C.; Lewis, G.D.; Vasan, R.S.; Carr, S.A.; Thadhani, R.; Gerszten, R.E.; Mootha, V.K. Metabolic profiling of the human response to a glucose challenge reveals distinct axes of insulin sensitivity. Mol. Syst. Biol. 2008, 4, 214. [Google Scholar]
- Pietilainen, K.H.; Naukkarinen, J.; Rissanen, A.; Saharinen, J.; Ellonen, P.; Keranen, H.; Suomalainen, A.; Gotz, A.; Suortti, T.; Yki-Jarvinen, H.; et al. lobal transcript profiles of fat in monozygotic twins discordant for BMI: Pathways behind acquired obesity. PLoS Med. 2008, 5, 472–483. [Google Scholar]
- Poulsen, M.; Mortensen, A.; Binderup, M.L.; Langkilde, S.; Markowski, J.; Dragsted, L.O. The effect of apple feeding on markers of colon carcinogenesis. Nutr. Cancer 2011, 63, 402–409. [Google Scholar] [CrossRef]
- Bergmeyer, H.U.; Gawahn, G.; Grassl, M. Methods of Enzymatic Analysis, 2nd ed; Academic Press Inc.: New York, NY, USA, 1974. [Google Scholar]
- Pete, M.J.; Ross, A.H.; Exton, J.H. Purification and Properties of Phospholipase-A(1) from Bovine Brain. J. Biol. Chem. 1994, 269, 19494–19500. [Google Scholar]
- Skov, T.; Bro, R. Solving fundamental problems in chromatographic analysis. Anal. Bioanal. Chem. 2008, 390, 281–285. [Google Scholar] [CrossRef]
- Savorani, F.; Tomasi, G.; Engelsen, S.B. icoshift: A versatile tool for the rapid alignment of 1D NMR spectra. J. Magn. Reson. 2010, 202, 190–202. [Google Scholar]
- Wold, S.; Esbensen, K.; Geladi, P. Principal Component Analysis. Chemom. Intell. Lab. Syst. 1987, 2, 37–52. [Google Scholar] [CrossRef]
- Wold, S.; Sjostrom, M.; Eriksson, L. PLS-regression: A basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Bijlsma, S.; Bobeldijk, I.; Verheij, E. R.; Ramaker, R.; Kochhar, S.; Macdonald, I. A.; van, O. B.; Smilde, A. K. Large-scale human metabolomics studies: A strategy for data (pre-) processing and validation. Anal. Chem. 2006, 78, 567–574. [Google Scholar]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. hMDB: The human metabolome database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gürdeniz, G.; Kristensen, M.; Skov, T.; Dragsted, L.O. The Effect of LC-MS Data Preprocessing Methods on the Selection of Plasma Biomarkers in Fed vs. Fasted Rats. Metabolites 2012, 2, 77-99. https://doi.org/10.3390/metabo2010077
Gürdeniz G, Kristensen M, Skov T, Dragsted LO. The Effect of LC-MS Data Preprocessing Methods on the Selection of Plasma Biomarkers in Fed vs. Fasted Rats. Metabolites. 2012; 2(1):77-99. https://doi.org/10.3390/metabo2010077
Chicago/Turabian StyleGürdeniz, Gözde, Mette Kristensen, Thomas Skov, and Lars O. Dragsted. 2012. "The Effect of LC-MS Data Preprocessing Methods on the Selection of Plasma Biomarkers in Fed vs. Fasted Rats" Metabolites 2, no. 1: 77-99. https://doi.org/10.3390/metabo2010077
APA StyleGürdeniz, G., Kristensen, M., Skov, T., & Dragsted, L. O. (2012). The Effect of LC-MS Data Preprocessing Methods on the Selection of Plasma Biomarkers in Fed vs. Fasted Rats. Metabolites, 2(1), 77-99. https://doi.org/10.3390/metabo2010077