2. Materials and Methods
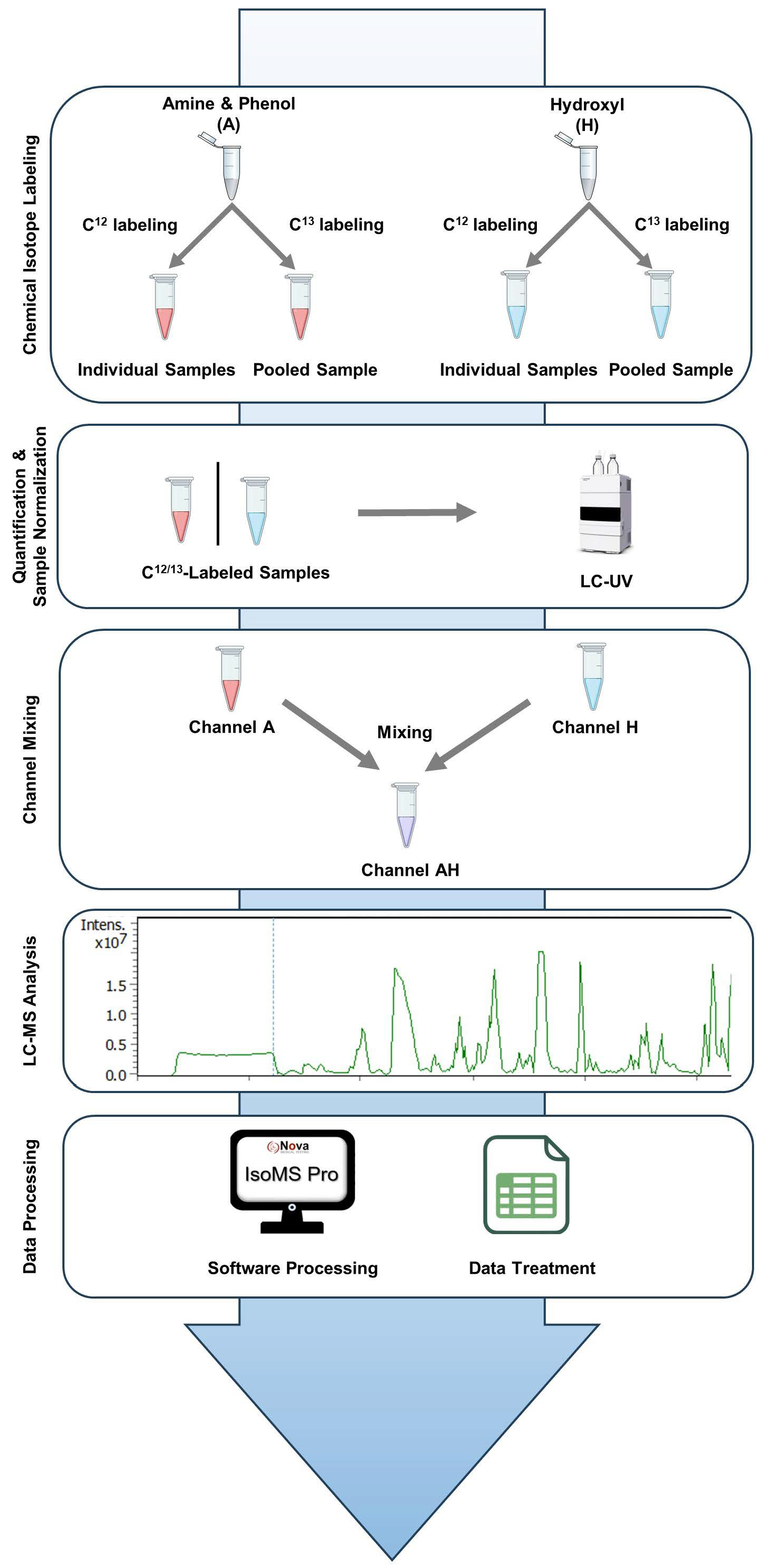
Study Workflow. The study workflow consists of five key steps, as illustrated in
Figure 1. Step 1 is chemical isotope labeling. Metabolites containing amine/phenol (A) and hydroxyl (H) groups were chemically tagged to enhance detection and quantitative measurement. Individual samples were labeled with
12C-Dansyl Chloride (
12C-DnsCl) (light labeling). A pooled sample, created by combining equal aliquots of all individual samples, was labeled with
13C-Dansyl Chloride (
13C-DnsCl) (heavy labeling). The heavy-labeled pooled sample served as an internal standard for all light-labeled samples, with equal amounts added to each sample based on quantification results (see below), forming a
12C/
13C mixture for LC-MS analysis. Step 2 involves sample quantification and normalization. Metabolome concentrations were measured using LC-UV and normalized to ensure uniformity for subsequent dilution and mixing. Step 3 is channel mixing. Equal parts of the amine/phenol (A) and hydroxyl (H) labeled samples were combined to create an AH mixture (AH) for combined analysis. Step 4 is LC-MS analysis. The A-, H-, and AH-labeled samples were analyzed by LC-MS. Metabolites were detected as peak pairs, distinguishing true metabolites from noise or unlabeled background signals. Step 5 is data processing. Raw LC-MS data were processed using IsoMS Pro (V1.4.0) software for peak pair detection, quantification, and metabolite identification. Further statistical analysis was performed in IsoMS Pro and Microsoft Excel (2016).
To evaluate the method’s performance, 16 urine samples and 15 serum samples were analyzed across individual channels (A and H) and the mixed channel (AH). A proof-of-concept study was conducted using 16 urine samples from two donors to further validate the approach’s feasibility for putative biomarker discovery.
Chemicals and Reagents. All chemicals and reagents, unless otherwise specified, were obtained from Sigma-Aldrich Canada (St. Louis, MO, USA). Chemical isotope labeling kits (CIL-4101-KT and CIL-4145-KT) were purchased from Nova Medical Testing Inc. (Edmonton, AB, Canada,
www.novamt.com, accessed on 3 October 2022). LC-MS-grade solvents, including water, acetonitrile (ACN), and methanol (MeOH), were procured from Thermo Fisher Scientific (Waltham, MA, USA) and Honeywell Research Chemicals (Charlotte, NC, USA).
Sample Pretreatment. Serum samples (30 μL) were mixed with 90 μL of pre-cooled methanol (MeOH), vortexed, and centrifuged at 10,000× g for 10 min to remove proteins. The supernatant was frozen at −20 °C for 1 h, re-centrifuged, and vacuum-dried. The residue was reconstituted with 25 μL of H2O for amine/phenol labeling or 25 μL of 3:1 ACN/H2O for hydroxyl labeling. Pre-filtered urine samples (12.5 μL) were thawed at 4 °C and centrifuged at 10,000× g for 10 min. For amine/phenol labeling, samples were diluted 4× with 37.5 μL of H2O. For hydroxyl labeling, samples were diluted with 37.5 μL of ACN to achieve a 3:1 ACN/H2O matrix. A 25 μL aliquot of each diluted sample was used for labeling.
Chemical Isotope Labeling. Chemical isotope labeling was performed following standard procedures provided in the CIL-4101-KT and CIL-4145-KT kits (Nova Medical Testing Inc., Edmonton, AB, Canada). Amine-/phenol-containing metabolites were labeled through a dansylation reaction, while hydroxyl-containing metabolites were labeled using a base-activated dansylation reaction.
Sample Normalization. Metabolite concentrations were quantified using a validated LC-UV method on an Agilent 1220 Infinity II HPLC system with a variable wavelength detector (VWD) [
19]. Mobile phase A was 0.1% formic acid in 5% ACN/H
2O, and mobile phase B was 0.1% formic acid in ACN. The LC gradient was set as follows: 0 min, 0% B; 0.01 min, 95% B; 2.5 min, 95% B; 3 min, 0% B; 6 min, 0% B. The flow rate was 0.45 mL/min. Metabolites were detected by measuring UV absorption at 338 nm. Metabolite concentrations were determined from the area under the curve, and equal amounts of
12C- and
13C-labeled samples were mixed for subsequent LC-MS analysis.
LC-MS Analysis. Labeled urine samples were analyzed using a Dionex UltiMate 3000 UHPLC system (Thermo Fisher Scientific, USA) coupled to a Maxis II QTOF mass spectrometer (Bruker Daltonics, Billerica, MA, USA) with an Eclipse Plus C18 column (2.1 × 100 mm, 1.8 μm, 95 Å, Agilent, Santa Clara, CA, USA). Mobile phase A was 0.1% formic acid in water, and mobile phase B was 0.1% formic acid in ACN. The LC gradient was set as follows: 0 min, 25% B; 10 min, 99% B; 15 min, 99% B; 15.1 min, 25% B; 18 min, 25% B. The flow rate was 400 µL/min, with a column temperature at 40 °C. MS data were acquired in positive ion mode at 1 Hz.
Similarly, serum samples were analyzed using an Agilent 1290 Infinities II LC system coupled with an Impact II QTOF mass spectrometer (Bruker Daltonics, Billerica, MA, USA), under identical conditions as for urine samples.
Data Processing and Metabolite Identification. Raw LC-MS data were processed using IsoMS Pro software, including peak pair detection, quantification, filtering, alignment, and imputation [
20]. Metabolite identification followed a three-tiered approach using the NovaMT Metabolite Databases 2.0 (Nova Medical Testing Inc., Canada). In tier 1, peak pairs matched against the labeled metabolite library (CIL Library) based on accurate mass and retention time as positive identification. In tier 2, remaining peak pairs matched to the linked identity (LI) library, containing 9000+ pathway-related metabolites with predicted retention times, generating high-confidence putative identifications. In tier 3, remaining peak pairs matched to the MyCompoundID (MCID) Library (
www.mycompoundid.org, accessed on 16 April 2024), which includes 8021 known endogenous metabolites in the zero-reaction library, 375,809 predicted metabolites in the one-reaction library, and 10,583,901 predicted metabolites in the two-reaction library. The resulting metabolite-intensity table was used for further statistical analysis.
Peak Pair Analysis. Peak pair detection was performed using IsoMS Pro, which identifies labeled metabolites through the detection of co-eluting 12C/13C-labeled ion pairs (i.e., peak pairs) that exhibit the expected mass difference of 2.0067 Da. Overlapping and unique peak pairs were determined by comparing m/z and retention time across different channels. Peak pairs were considered overlapping if both m/z and retention time matched within a defined tolerance window. All comparisons were performed after data alignment and filtering. Post-channel mixing peak pair loss was assessed by comparing LC-MS spectra between individual channels (A or H) and the AH mixture at the same retention time and m/z region. First, a peak pair that was clearly detected in the individual channel analysis and matched the expected isotopic spacing of 2.0067 Da between 12C/13C-labeled peak pairs was selected. The relevant m/z window was then enlarged to enhance clarity of peak shape, spacing, and intensity. The same retention time and m/z region were examined in the AH mixture. A peak pair was considered lost if absent or below the detection threshold in the AH mixture, but clearly present in the single channel.
Statistical Analysis. Metabolite-intensity data were analyzed using IsoMS Pro for univariate and multivariate analyses. Significant metabolites were determined using volcano plots with the following criteria: fold change > 1.2 or <0.83, p-value < 0.05, and q-value (FDR-adjusted p-value) < 0.25. All three criteria were used to define significant metabolites in volcano plot analyses. The relaxed q-value threshold served to reduce the risk of false negatives and avoid overlooking potentially meaningful metabolites, while the application of fold-change and p-value criteria further refined the selection of significant features. Additional analyses, such as Venn diagrams, were performed using Microsoft Excel.
3. Results and Discussion
Peak Pair Detectability. Peak pair detection in the single-channel analysis of amine/phenol submetabolome (A) and hydroxyl submetabolome (H) is summarized in
Figure 2 for urine (
Figure 2A–C) and serum samples (
Figure 2D–F). Further information is provided in
Supplementary Tables S1 and S2. In urine samples, 3502 peak pairs were detected in the A channel and 4041 were detected in the H channel (
Figure 2A). Less than 25.0% of the detected peak pairs overlapped between the two channels, highlighting the specificity of the chemical labeling techniques employed and the need for a four-channel labeling strategy to achieve comprehensive metabolite coverage.
The AH mixture captured 2224 peak pairs from the A channel (63.5% coverage,
Figure 2B) and 2558 peak pairs (63.3% coverage,
Figure 2C) from the H channel. Similar results were observed for serum samples: 2751 peak pairs were detected in the A channel and 3744 in the H channel, with 80% of peak pairs being unique to each channel (
Figure 2D). The AH mixture retained 1226 peak pairs from the A channel (46.1% coverage,
Figure 2E) and 2470 from the H channel (66.0% coverage,
Figure 2F).
Notably, in urine samples, the AH mixture captured a higher percentage of peak pairs from the A channel compared to the H channel, whereas in serum samples, the opposite trend was observed. This observation suggests differences in ion suppression effects between urine and serum, emphasizing that ion suppression varies with sample type. These findings indicate the importance of understanding ion suppression effects in metabolomic analyses to ensure accurate and comprehensive metabolite detection across different sample types.
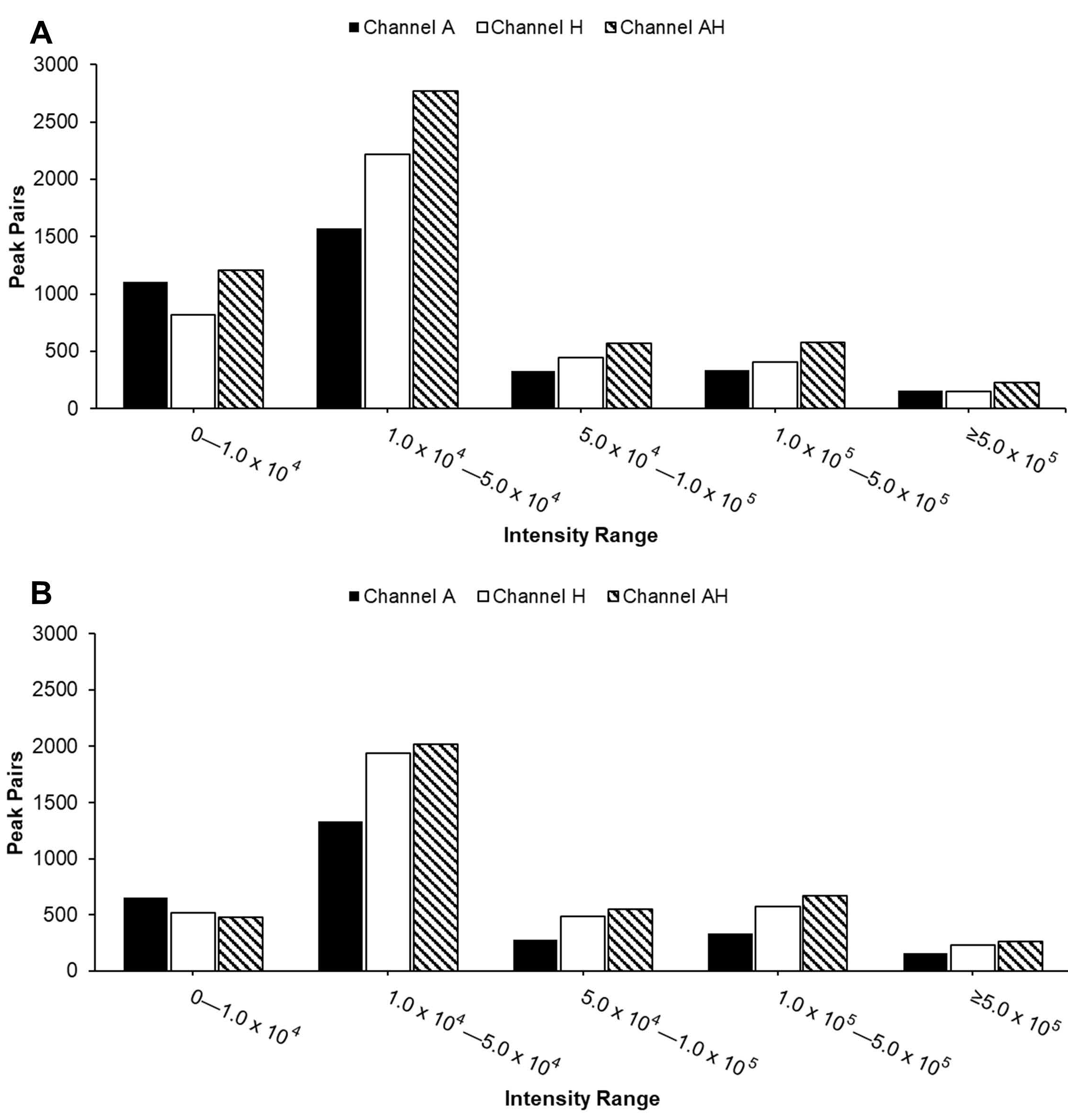
Ion Suppression Effect. To further investigate ion suppression, we calculated the percentage of peak pair loss in the AH channel, as illustrated in
Figure 3. Peak pair loss was defined as follows: Peak pair loss = (Number of peak pairs detected in single channel but absent in AH channel)/(Number of peak pairs in single channel). Higher values indicate greater loss of peak pairs during channel mixing. The results showed that peak pairs at lower intensities were more susceptible to loss due to ion suppression across both sample types.
For urine samples, peak pair losses of the A channel were lower compared to the H channel in most of the intensity ranges, except for the lowest range (
Figure 3A), suggesting that amine-/phenol-containing metabolites were better preserved during mixing. In contrast, serum samples displayed higher peak pair loss in the A channel compared to the H channel (
Figure 3B), indicating a distinct detection preference depending on sample type and intensity region.
Compared to individual channels, the AH mixture consistently detected the highest number of peak pairs across nearly all intensity ranges (
Figure 4), as expected due to the combination of metabolites from both A and H channels. Most peak pairs in both urine and serum samples were detected in the two lowest intensity ranges. The reduced peak pair coverage observed in
Figure 2 can be attributed to significant ion suppression effects seen in
Figure 3. While the overall distribution of peak pairs was similar between sample types, urine samples exhibited a higher abundance of peak pairs in the lowest intensity range, reflecting their intrinsic chemical complexity.
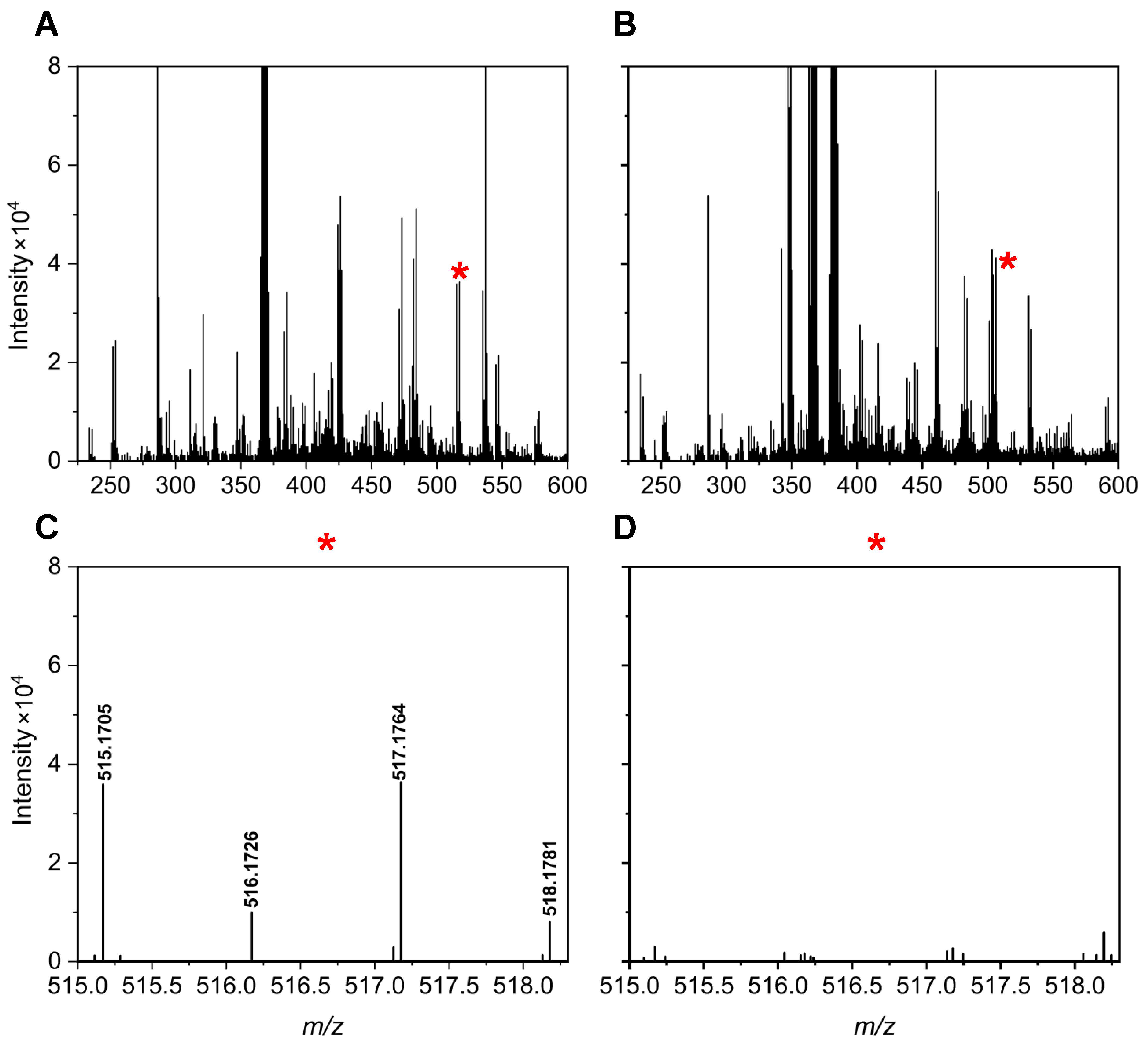
Figure 5 and
Figure 6 illustrate specific examples of peak pairs lost post-mixing. For instance, a peak pair with an
m/
z value of 515.1705/517.1764, initially detected in channel A, disappeared in the mixed AH channel (
Figure 5). Similarly, a peak pair with an
m/
z value of 424.1205/426.1268 found in channel H was absent after mixing (
Figure 6).
One potential strategy to address ion suppression, particularly for low-intensity metabolites, is to incorporate sample fractionation during the preparation steps. This process separates complex samples into simpler fractions, thereby reducing matrix effects and enhancing detection sensitivity [
21]. Additionally, LC separation conditions can be further optimized to match the composition of the two-channel mixture, as improved chromatographic resolution allows for better separation of metabolites, aiding in ionization efficiency. Alternatively, if comprehensive profiling is a priority, the original four-channel labeling strategy may be more appropriate. CIL significantly enhances the signal intensity of labeled metabolites, improving detectability, particularly for low-abundance compounds [
22]. Overall, ion suppression is mainly caused by the inherent complexity of samples like urine and serum. There is an unavoidable trade-off between throughput and sensitivity, but channel mixing is customized for high-throughput analysis while accepting a minor reduction in metabolome coverage.
Metabolite Identification. The number of highly confident identified metabolites (tier 1 and tier 2) between single channels (A and H) and the AH mixture were compared, as shown in
Figure 7. For urine samples, the A channel identified 714 metabolites, comprising 245 tier 1 and 469 tier 2 metabolites (
Figure 7A). The H channel identified 393 metabolites, including 67 tier 1 and 326 tier 2 metabolites (
Figure 7B). The AH combined channel covered 90.5% of the metabolites identified in the A channel and 91.6% of those identified in the H channel, indicating efficient retention of key metabolites. Similar results were observed for serum samples. Of the 657 metabolites identified in the A channel (325 tier 1 and 332 tier 2,
Figure 7C), the AH combined channel captured 524 metabolites (79.8% coverage). For the H channel, 415 out of 490 metabolites (84.7%) were retained in the AH mixture (
Figure 7D).
The higher coverage percentage of identified metabolites compared to the detected peak pairs suggests that, although some metabolite candidates may be lost in the combined channel analysis, the most critical information, i.e., high-confidence identified metabolites, is largely preserved. This highlights the robustness of the AH combined channel for metabolite identification, ensuring effective data integration without significant loss of essential findings.
The observed loss of coverage in the AH combined channel reflects the trade-off between analytical throughput and metabolite coverage; while some reduction in coverage was noted, a significantly higher throughput was achieved while maintaining an acceptable level of metabolite detection. Additionally, as shown in
Figure 3, the coverage loss primarily occurred among peak pairs with low signal intensity, which are features that typically exhibit lower detection consistency and quantification accuracy [
3].
Proof of Concept Study. To evaluate the feasibility of our method, we analyzed 16 urine samples from two distinct donors. Each donor contributed eight samples collected over three days at morning, noon, and evening intervals. These donors represented two unique population groups, providing valuable insights into the method’s practicality for real-world research, such as putative biomarker discovery.
Using volcano plots as a univariate analysis tool, we identified metabolites with significant differences, serving as potential biomarker candidates to differentiate samples between the two donors. Across channels A, H, and AH, we identified 1630, 1512, and 2569 significant metabolites, respectively (
Figure 8A–C). A Venn diagram of highly confident identified metabolites (tier 1 and tier 2) (
Figure 8D) revealed that the AH channel captured 63.7% and 57.4% of the significantly altered metabolites identified in the A and H channels, respectively. Despite detecting fewer metabolites overall compared to the combined totals of the A and H channels, the AH channel identified a substantial number of significant metabolites while reducing analysis time by half. Further information is provided in
Supplemental Tables S3–S5.
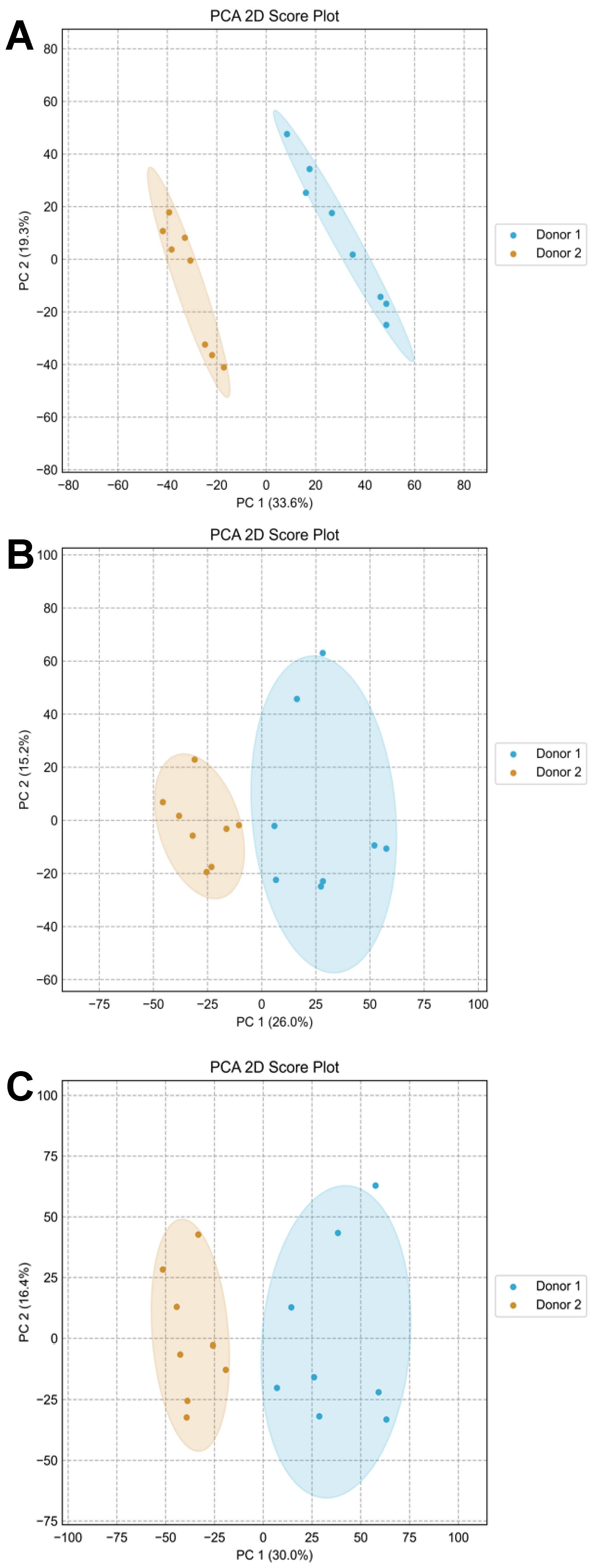
Principal Component Analysis (PCA) was conducted to visually examine the distribution of the 16 samples. The PCA for amine-/phenol-containing metabolites showed clear separation between the two donors, indicating distinct metabolite compositions (
Figure 9A). In contrast, the PCA for hydroxyl-containing metabolites displayed slightly overlapping clusters, suggesting limited compositional differences for this metabolite subset (
Figure 9B). Notably, the PCA for the AH mixture effectively integrated the unique characteristics from both A and H channels, resulting in clear separation and demonstrating successful data integration from both submetabolomes (
Figure 9C).
Importantly, the eight samples collected from each donor at different times clustered closely together, reflecting the consistency and stability of each donor’s unique metabolite composition. This underscores the reliability of the CIL approach in preserving sample integrity over time. Additionally, despite the modest loss in coverage, both univariate and multivariate analyses demonstrate that the AH channel is still capable of capturing key metabolic differences between sample groups. This indicates that the loss of metabolite coverage through channel mixing does not significantly compromise the interpretability or validity of results. Moreover, the method’s ability to accurately capture an individual’s metabolic fingerprint across multiple sampling instances highlights its potential for biomarker exploration and longitudinal studies.
Future Directions. The two-channel mixing method designed was created to preserve group-wise comparability while improving throughput, rather than to optimize for absolute sensitivity or dynamic range. As a result, our study was focused on relative quantification across biological samples, not absolute quantification. We propose standard-based validation for evaluating sensitivity, linearity, and dynamic range to be conducted as a future direction [
23].
The proposed two-channel mixing method reduces LC-MS runtime by half, as only one injection is needed due to the pre-analysis combination of amine-/phenol-labeled and hydroxyl-labeled samples. This reduction in time required per sample allows for substantial improvements in throughput that would positively impact studies in which thousands of samples must be processed, such as population-scale or longitudinal studies [
24]. Additionally, the reduced runtime minimizes instrument downtime and required maintenance, allowing for continuous large-scale sample acquisition. Channel mixing after labeling the samples creates a modular workflow, supporting future automation [
25]. This modular workflow allows our approach to be compatible with standardized, parallel sample preparation workflows, resulting in increased scalability and reproducibility across large datasets.
In this study, the two-channel approach was applied to human urine and serum samples, two biologically distinct matrices that differ in protein content, salt load, and metabolite complexity [
26]. The results showed that the majority of high-confidence metabolites identified in individual channels were retained after mixing (>90% retained in AH mixture from urine A and H channels; >80% retained in AH mixture from serum A and H channels). Due to these results, we anticipate that this method can be applied to other complex sample types such as saliva, tissue, or cell lysate with only minor protocol adjustments.
To address current limitations related to sample types and metabolite coverage, future studies will focus on extending the two-channel mixing strategy to more complex biological matrices, such as tissues and cultured cells, which often require additional pretreatment steps [
27]. Evaluating the method’s performance across diverse sample types will help establish its robustness and generalizability. Additionally, while the two-channel approach captures a broad range of metabolomic features, specific one-channel analyses or targeted validation workflows may be employed in parallel to confirm key findings or to provide enhanced coverage of certain metabolite classes that may be underrepresented. These combined strategies will help to further improve the analytical depth and applicability of the method in both exploratory and hypothesis-driven studies.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}