
Metabolic Changes in Zebrafish Larvae Infected with Mycobacterium marinum: A Widely Targeted Metabolomic Analysis


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Zebrafish Cultivation
2.2. Zebrafish Embryo Infection and Sample Collection
2.3. Widely Targeted Metabolomics and Data Analysis
2.3.1. Pre-Processing
2.3.2. LC-MS/MS Analytical Conditions
2.3.3. Data Analysis
3. Results
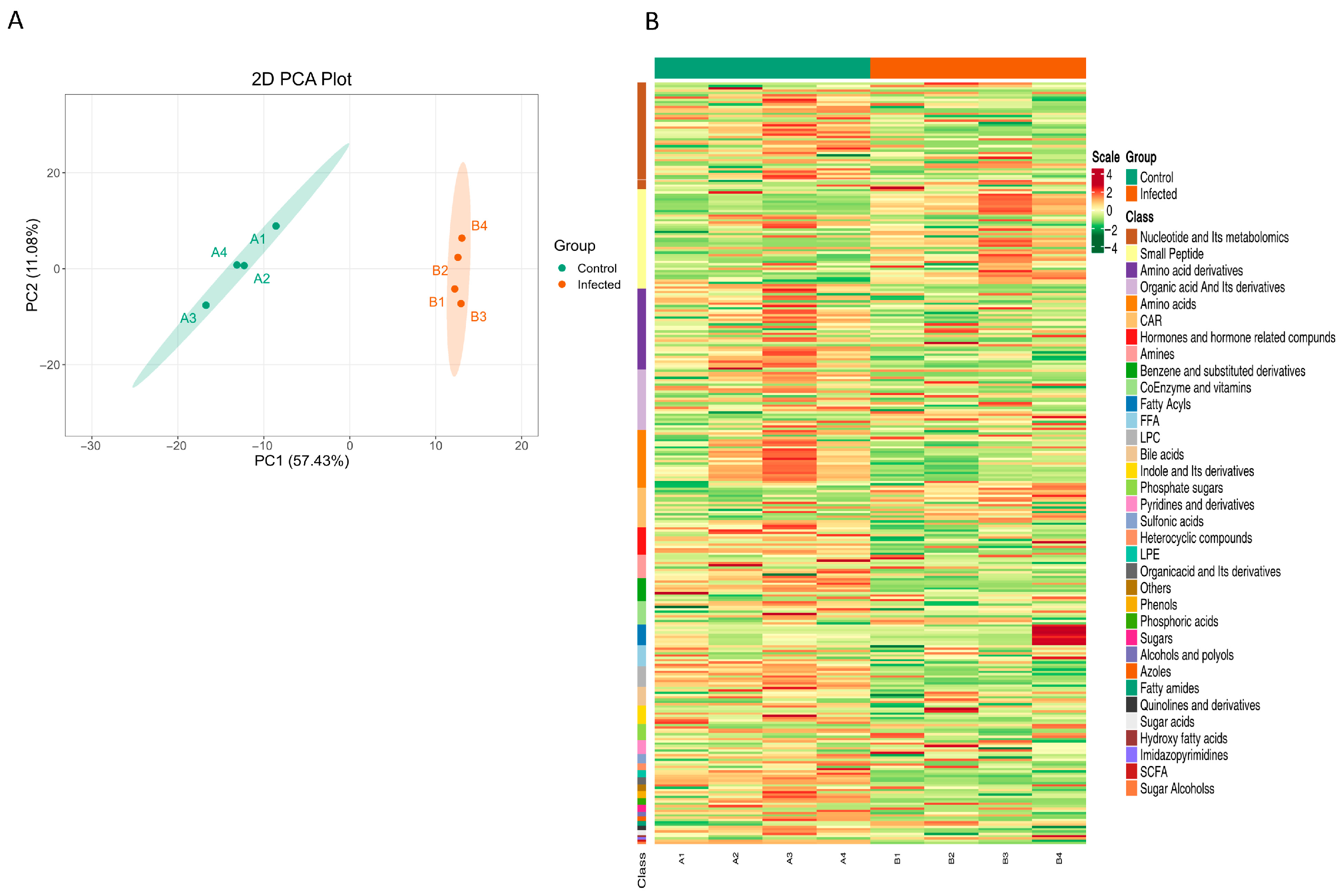
3.1. Metabolic Profile Changes in Zebrafish Larvae upon Infection
3.2. Screening of Differential Metabolites
3.3. KEGG Functional Annotation and Metabolic Pathway Analysis of Differential Metabolites
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, J.F. Mycobacterium tuberculosis and the cause of consumption: From discovery to fact. Am. J. Respir. Crit. Care Med. 2004, 169, 1086–1088. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2024. Available online: https://www.who.int/tb/publications/global_report/en/ (accessed on 26 May 2025).
- van Leeuwen, L.M.; van der Sar, A.M.; Bitter, W. Animal models of tuberculosis: Zebrafish. Cold Spring Harb. Perspect. Med. 2014, 5, a018580. [Google Scholar] [CrossRef]
- Lam, S.H.; Chua, H.L.; Gong, Z.; Lam, T.J.; Sin, Y.M. Development and maturation of the immune system in zebrafish, Danio rerio:: A gene expression profiling, in situ hybridization and immunological study. Dev. Comp. Immunol. 2004, 28, 9–28. [Google Scholar] [CrossRef]
- Cronan, M.R.; Tobin, D.M. Fit for consumption: Zebrafish as a model for tuberculosis. Dis. Models Mech. 2014, 7, 777–784. [Google Scholar] [CrossRef]
- Stinear, T.P.; Seemann, T.; Harrison, P.F.; Jenkin, G.A.; Davies, J.K.; Johnson, P.D.R.; Abdellah, Z.; Arrowsmith, C.; Chillingworth, T.; Churcher, C.; et al. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 2008, 18, 729–741. [Google Scholar] [CrossRef]
- Parikka, M.; Hammaren, M.M.; Harjula, S.-K.E.; Halfpenny, N.J.A.; Oksanen, K.E.; Lahtinen, M.J.; Pajula, E.T.; Iivanainen, A.; Pesu, M.; Ramet, M. Mycobacterium marinum Causes a Latent Infection that Can Be Reactivated by Gamma Irradiation in Adult Zebrafish. PLoS Pathog. 2012, 8, e1002944. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Lee, K.-C.; Curreem, S.O.T.; Chow, W.-N.; To, K.K.W.; Hung, I.F.N.; Ho, D.T.Y.; Sridhar, S.; Li, I.W.S.; Ding, V.S.Y.; et al. Metabolomic Profiling of Plasma from Patients with Tuberculosis by Use of Untargeted Mass Spectrometry Reveals Novel Biomarkers for Diagnosis. J. Clin. Microbiol. 2015, 53, 3750–3759. [Google Scholar] [CrossRef]
- Weiner, J., 3rd; Parida, S.K.; Maertzdorf, J.; Black, G.F.; Repsilber, D.; Telaar, A.; Mohney, R.P.; Arndt-Sullivan, C.; Ganoza, C.A.; Fae, K.C.; et al. Biomarkers of inflammation, immunosuppression and stress with active disease are revealed by metabolomic profiling of tuberculosis patients. PLoS ONE 2012, 7, e40221. [Google Scholar] [CrossRef]
- Collins, J.M.; Bobosha, K.; Narayanan, N.; Gandhi, N.R.; Day, C.L.; Rengarajan, J.; Kempker, R.R.; Lau, M.S.Y.; Nellis, M.; Tukvadze, N.; et al. A plasma metabolic signature to diagnose pulmonary tuberculosis and monitor treatment response. J. Infect. Dis. 2025, jiaf240. [Google Scholar] [CrossRef]
- Yu, J.; Yuan, J.; Liu, Z.; Ye, H.; Lin, M.; Ma, L.; Liu, R.; Ding, W.; Li, L.; Ma, T.; et al. Combined urine proteomics and metabolomics analysis for the diagnosis of pulmonary tuberculosis. Clin. Proteom. 2024, 21, 66. [Google Scholar] [CrossRef]
- Sa, Y.; Ding, S.; Zhang, Y.; Wang, W.; Wilson, G.; Ma, F.; Zhang, W.; Ma, X. Integrating untargeted and targeted LC-MS-based metabolomics to identify the serum metabolite biomarkers for tuberculosis. Biomed. Chromatogr. 2024, 38, e5998. [Google Scholar] [CrossRef]
- Ding, Y.; Raterink, R.J.; Marin-Juez, R.; Veneman, W.J.; Egbers, K.; van den Eeden, S.; Haks, M.C.; Joosten, S.A.; Ottenhoff, T.H.M.; Harms, A.C.; et al. Tuberculosis causes highly conserved metabolic changes in human patients, mycobacteria-infected mice and zebrafish larvae. Sci. Rep. 2020, 10, 11635. [Google Scholar] [CrossRef]
- Parveen, S.; Shen, J.; Lun, S.; Zhao, L.; Alt, J.; Koleske, B.; Leone, R.D.; Rais, R.; Powell, J.D.; Murphy, J.R.; et al. Glutamine metabolism inhibition has dual immunomodulatory and antibacterial activities against Mycobacterium tuberculosis. Nat. Commun. 2023, 14, 7427. [Google Scholar] [CrossRef]
- Liu, X.; Yang, M.; Xu, P.; Du, M.; Li, S.; Shi, J.; Li, Q.; Yuan, J.; Pang, Y. Kynurenine-AhR reduces T-cell infiltration and induces a delayed T-cell immune response by suppressing the STAT1-CXCL9/CXCL10 axis in tuberculosis. Cell. Mol. Immunol. 2024, 21, 1426–1440. [Google Scholar] [CrossRef]
- Niu, L.; Luo, G.; Liang, R.; Qiu, C.; Yang, J.; Xie, L.; Zhang, K.; Tian, Y.; Wang, D.; Song, S.; et al. Negative Regulator Nlrc3-like Maintain the Balanced Innate Immune Response During Mycobacterial Infection in Zebrafish. Front. Immunol. 2022, 13, 893611. [Google Scholar] [CrossRef]
- Vivekaa, A.; Nellore, J.; Sunkar, S. Zebrafish metabolomics: A comprehensive approach to understanding health and disease. Funct. Integr. Genom. 2025, 25, 110. [Google Scholar] [CrossRef]
- Zhang, K.; Mishra, A.; Jagannath, C. New insight into arginine and tryptophan metabolism in macrophage activation during tuberculosis. Front. Immunol. 2024, 15, 1363938. [Google Scholar] [CrossRef]
- Amalia, F.; Syamsunarno, M.; Triatin, R.D.; Fatimah, S.N.; Chaidir, L.; Achmad, T.H. The Role of Amino Acids in Tuberculosis Infection: A Literature Review. Metabolites 2022, 12, 933. [Google Scholar] [CrossRef]
- Collins, J.M.; Siddiqa, A.; Jones, D.P.; Liu, K.; Kempker, R.R.; Nizam, A.; Shah, N.S.; Ismail, N.; Ouma, S.G.; Tukvadze, N.; et al. Tryptophan catabolism reflects disease activity in human tuberculosis. JCI Insight 2020, 5, e137131. [Google Scholar] [CrossRef]
- Fadhilah, F.; Indrati, A.R.; Dewi, S.; Santoso, P. The Kynurenine/Tryptophan Ratio as a Promising Metabolomic Biomarker for Diagnosing the Spectrum of Tuberculosis Infection and Disease. Int. J. Gen. Med. 2023, 16, 5587–5595. [Google Scholar] [CrossRef]
- Ardiansyah, E.; Avila-Pacheco, J.; Nhat, L.T.H.; Dian, S.; Vinh, D.N.; Hai, H.T.; Bullock, K.; Alisjahbana, B.; Netea, M.G.; Estiasari, R.; et al. Tryptophan metabolism determines outcome in tuberculous meningitis: A targeted metabolomic analysis. Elife 2023, 12, e85307. [Google Scholar] [CrossRef]
- Wang, K.; Chen, T.L.; Zhang, X.X.; Cao, J.B.; Wang, P.; Wang, M.; Du, J.L.; Mu, Y.; Tao, R. Unveiling tryptophan dynamics and functions across model organisms via quantitative imaging. BMC Biol. 2024, 22, 258. [Google Scholar] [CrossRef]
- Marszalek-Grabska, M.; Turska-Kozlowska, M.; Kaczorek-Lukowska, E.; Wicha-Komsta, K.; Turski, W.A.; Siwicki, A.K.; Gawel, K. The Effects of Kynurenic Acid in Zebrafish Embryos and Adult Rainbow Trout. Biomolecules 2024, 14, 1148. [Google Scholar] [CrossRef]
- Brum, M.; Nieberler, M.; Kehrwald, C.; Knopf, K.; Brunkhorst-Kanaan, N.; Etyemez, S.; Allers, K.A.; Bittner, R.A.; Slattery, D.A.; McNeill, R.V.; et al. Phase-and disorder-specific differences in peripheral metabolites of the kynurenine pathway in major depression, bipolar affective disorder and schizophrenia. World J. Biol. Psychiatry 2023, 24, 564–577. [Google Scholar] [CrossRef]
- Alp, H.H.; Kurhan, F.; Akbay, H.I. Predictive value of kynurenine pathway metabolites in the severity of patients with obsessive-compulsive disorder. Psychiatry Clin. Neurosci. 2025; online ahead of print. [Google Scholar]
- Gleeson, L.E.; Sheedy, F.J.; Palsson-McDermott, E.M.; Triglia, D.; O’Leary, S.M.; O’Sullivan, M.P.; O’Neill, L.A.; Keane, J. Cutting Edge: Mycobacterium tuberculosis Induces Aerobic Glycolysis in Human Alveolar Macrophages That Is Required for Control of Intracellular Bacillary Replication. J. Immunol. 2016, 196, 2444–2449. [Google Scholar] [CrossRef]
- Osada-Oka, M.; Goda, N.; Saiga, H.; Yamamoto, M.; Takeda, K.; Ozeki, Y.; Yamaguchi, T.; Soga, T.; Tateishi, Y.; Miura, K.; et al. Metabolic adaptation to glycolysis is a basic defense mechanism of macrophages for Mycobacterium tuberculosis infection. Int. Immunol. 2019, 31, 781–793. [Google Scholar] [CrossRef]
- Maoldomhnaigh, C.Ó.; Cox, D.J.; Phelan, J.J.; Mitermite, M.; Murphy, D.M.; Leisching, G.; Thong, L.; O’Leary, S.M.; Gogan, K.M.; McQuaid, K.; et al. Lactate Alters Metabolism in Human Macrophages and Improves Their Ability to Kill Mycobacterium tuberculosis. Front. Immunol. 2021, 12, 663695. [Google Scholar] [CrossRef]
- Huang, Y.; Li, R.; Chen, S.; Wang, Q.; Han, Z.; Liang, Y.; Liang, Y.; Li, Q.; Lin, L.; Wen, Q.; et al. Monocarboxylate transporter 4 facilitates Mycobacterium tuberculosis survival through NF-kappaB p65-mediated interleukin-10 production. iScience 2024, 27, 110238. [Google Scholar] [CrossRef]
- Yin, J.; Forn-Cuni, G.; Surendran, A.M.; Lopes-Bastos, B.; Pouliopoulou, N.; Jager, M.J.; Le Devedec, S.E.; Chen, Q.; Snaar-Jagalska, B.E. Lactate secreted by glycolytic conjunctival melanoma cells attracts and polarizes macrophages to drive angiogenesis in zebrafish xenografts. Angiogenesis 2024, 27, 703–717. [Google Scholar] [CrossRef]
- Zhou, R.; Ding, R.C.; Yu, Q.; Qiu, C.Z.; Zhang, H.Y.; Yin, Z.J.; Ren, D.L. Metformin Attenuates Neutrophil Recruitment through the H3K18 Lactylation/Reactive Oxygen Species Pathway in Zebrafish. Antioxidants 2024, 13, 176. [Google Scholar] [CrossRef]
- Roca, F.J.; Whitworth, L.J.; Prag, H.A.; Murphy, M.P.; Ramakrishnan, L. Tumor necrosis factor induces pathogenic mitochondrial ROS in tuberculosis through reverse electron transport. Science 2022, 376, eabh2841. [Google Scholar] [CrossRef]
- Scott, C.A.; Carney, T.J.; Amaya, E. Aerobic glycolysis is important for zebrafish larval wound closure and tail regeneration. Wound Repair. Regen. 2022, 30, 665–680. [Google Scholar] [CrossRef]
- Hoppel, C. The role of carnitine in normal and altered fatty acid metabolism. Am. J. Kidney Dis. 2003, 41, S4–S12. [Google Scholar] [CrossRef]
- Ito, S.; Taguchi, K.; Kodama, G.; Kubo, S.; Moriyama, T.; Yamashita, Y.; Yokota, Y.; Nakayama, Y.; Kaida, Y.; Shinohara, M.; et al. Involvement of impaired carnitine-induced fatty acid oxidation in experimental and human diabetic kidney disease. JCI Insight 2025, e179362. [Google Scholar] [CrossRef]
- Sankar, P.; Ramos, R.B.; Corro, J.; Mishra, L.K.; Nafiz, T.N.; Bhargavi, G.; Saqib, M.; Poswayo, S.K.L.; Parihar, S.P.; Cai, Y.; et al. Fatty acid metabolism in neutrophils promotes lung damage and bacterial replication during tuberculosis. PLoS Pathog. 2024, 20, e1012188. [Google Scholar] [CrossRef]
- Bender, M.J.; McPherson, A.C.; Phelps, C.M.; Pandey, S.P.; Laughlin, C.R.; Shapira, J.H.; Medina Sanchez, L.; Rana, M.; Richie, T.G.; Mims, T.S.; et al. Dietary tryptophan metabolite released by intratumoral Lactobacillus reuteri facilitates immune checkpoint inhibitor treatment. Cell 2023, 186, 1846–1862 e1826. [Google Scholar] [CrossRef]
- Strassman, M.; Ceci, L.N. Enzymatic formation of alpha-isopropylmalic acid, an intermediate in leucine biosynthesis. J. Biol. Chem. 1963, 238, 2445–2452. [Google Scholar] [CrossRef]
- Ricciutelli, M.; Bartolucci, G.; Campana, R.; Salucci, S.; Benedetti, S.; Caprioli, G.; Maggi, F.; Sagratini, G.; Vittori, S.; Lucarini, S. Quantification of 2- and 3-isopropylmalic acids in forty Italian wines by UHPLC-MS/MS triple quadrupole and evaluation of their antimicrobial, antioxidant activities and biocompatibility. Food Chem. 2020, 321, 126726. [Google Scholar] [CrossRef]
- Ricciutelli, M.; Moretti, S.; Galarini, R.; Sagratini, G.; Mari, M.; Lucarini, S.; Vittori, S.; Caprioli, G. Identification and quantification of new isomers of isopropyl-malic acid in wine by LC-IT and LC-Q-Orbitrap. Food Chem. 2019, 294, 390–396. [Google Scholar] [CrossRef]
- Yan, H.; Liu, Y.; Li, X.; Yu, B.; He, J.; Mao, X.; Yu, J.; Huang, Z.; Luo, Y.; Luo, J.; et al. Leucine alleviates cytokine storm syndrome by regulating macrophage polarization via the mTORC1/LXRalpha signaling pathway. Elife 2024, 12, RP89750. [Google Scholar] [CrossRef]
- Ananieva, E.A.; Powell, J.D.; Hutson, S.M. Leucine Metabolism in T Cell Activation: mTOR Signaling and Beyond. Adv. Nutr. 2016, 7, 798S–805S. [Google Scholar] [CrossRef]
- Cai, J.; Sun, L.; Gonzalez, F.J. Gut microbiota-derived bile acids in intestinal immunity, inflammation, and tumorigenesis. Cell Host Microbe 2022, 30, 289–300. [Google Scholar] [CrossRef]
- do Nascimento, P.G.; Lemos, T.L.; Almeida, M.C.; de Souza, J.M.; Bizerra, A.M.; Santiago, G.M.; da Costa, J.G.; Coutinho, H.D. Lithocholic acid and derivatives: Antibacterial activity. Steroids 2015, 104, 8–15. [Google Scholar] [CrossRef]
- Maji, A.; Misra, R.; Dhakan, D.B.; Gupta, V.; Mahato, N.K.; Saxena, R.; Mittal, P.; Thukral, N.; Sharma, E.; Singh, A.; et al. Gut microbiome contributes to impairment of immunity in pulmonary tuberculosis patients by alteration of butyrate and propionate producers. Environ. Microbiol. 2018, 20, 402–419. [Google Scholar] [CrossRef]
- Zhang, H.; Xue, M.; He, X.; Sun, L.; He, Q.; Wang, Y.; Jin, J. Altered intestinal microbiota and fecal metabolites in patients with latent and active pulmonary tuberculosis. Tuberculosis 2024, 149, 102577. [Google Scholar] [CrossRef]
- Wang, S.; Yang, L.; Hu, H.; Lv, L.; Ji, Z.; Zhao, Y.; Zhang, H.; Xu, M.; Fang, R.; Zheng, L.; et al. Characteristic gut microbiota and metabolic changes in patients with pulmonary tuberculosis. Microb. Biotechnol. 2022, 15, 262–275. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, G.; Li, G.; Wang, W.; Ge, Z.; Yang, Y.; He, X.; Liu, Z.; Zhang, Z.; Mai, Q.; et al. Ifnar gene variants influence gut microbial production of palmitoleic acid and host immune responses to tuberculosis. Nat. Metab. 2022, 4, 359–373. [Google Scholar] [CrossRef]
- Yang, B.; Guo, X.; Shi, C.; Liu, G.; Qin, X.; Chen, S.; Gan, L.; Liang, D.; Shao, K.; Xu, R.; et al. Alterations in purine and pyrimidine metabolism associated with latent tuberculosis infection: Insights from gut microbiome and metabolomics analyses. mSystems 2024, 9, e0081224. [Google Scholar] [CrossRef]
- Shi, K.X.; Wu, Y.K.; Tang, B.K.; Zhao, G.P.; Lyu, L.D. Housecleaning of pyrimidine nucleotide pool coordinates metabolic adaptation of nongrowing Mycobacterium tuberculosis. Emerg. Microbes Infect. 2019, 8, 40–44. [Google Scholar] [CrossRef]
- Brewer, W.J.; Xet-Mull, A.M.; Yu, A.; Sweeney, M.I.; Walton, E.M.; Tobin, D.M. Macrophage NFATC2 mediates angiogenic signaling during mycobacterial infection. Cell Rep. 2022, 41, 111817. [Google Scholar] [CrossRef]
- Pagan, A.J.; Lee, L.J.; Edwards-Hicks, J.; Moens, C.B.; Tobin, D.M.; Busch-Nentwich, E.M.; Pearce, E.L.; Ramakrishnan, L. mTOR-regulated mitochondrial metabolism limits mycobacterium-induced cytotoxicity. Cell 2022, 185, 3720–3738.e3713. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, M.J.; Venketaraman, V. Advances in Host-Pathogen Interactions in Tuberculosis: Emerging Strategies for Therapeutic Intervention. Int. J. Mol. Sci. 2025, 26, 1621. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sima, C.; Zhang, Q.; Yu, X.; Yan, B.; Zhang, S. Metabolic Changes in Zebrafish Larvae Infected with Mycobacterium marinum: A Widely Targeted Metabolomic Analysis. Metabolites 2025, 15, 449. https://doi.org/10.3390/metabo15070449
Sima C, Zhang Q, Yu X, Yan B, Zhang S. Metabolic Changes in Zebrafish Larvae Infected with Mycobacterium marinum: A Widely Targeted Metabolomic Analysis. Metabolites. 2025; 15(7):449. https://doi.org/10.3390/metabo15070449
Chicago/Turabian StyleSima, Chongyuan, Qifan Zhang, Xiaoli Yu, Bo Yan, and Shulin Zhang. 2025. "Metabolic Changes in Zebrafish Larvae Infected with Mycobacterium marinum: A Widely Targeted Metabolomic Analysis" Metabolites 15, no. 7: 449. https://doi.org/10.3390/metabo15070449
APA StyleSima, C., Zhang, Q., Yu, X., Yan, B., & Zhang, S. (2025). Metabolic Changes in Zebrafish Larvae Infected with Mycobacterium marinum: A Widely Targeted Metabolomic Analysis. Metabolites, 15(7), 449. https://doi.org/10.3390/metabo15070449