
A Systems Hypothesis of Lipopolysaccharide-Induced Vitamin Transport Suppression and Metabolic Reprogramming in Autism Spectrum Disorders: An Open Call for Validation and Therapeutic Translation


Abstract

1. Introduction
2. Methods
2.1. Transcriptomic Dataset Acquisition
2.2. PM3 Gene Model Construction
2.3. Differential Expression and Divergence Metrics
2.4. Machine Learning and Gene-Level Anomaly Detection
2.5. Software and Visualization Tools
3. Results
3.1. Divergence of Metabolic Gene Expression in ASD Cortex
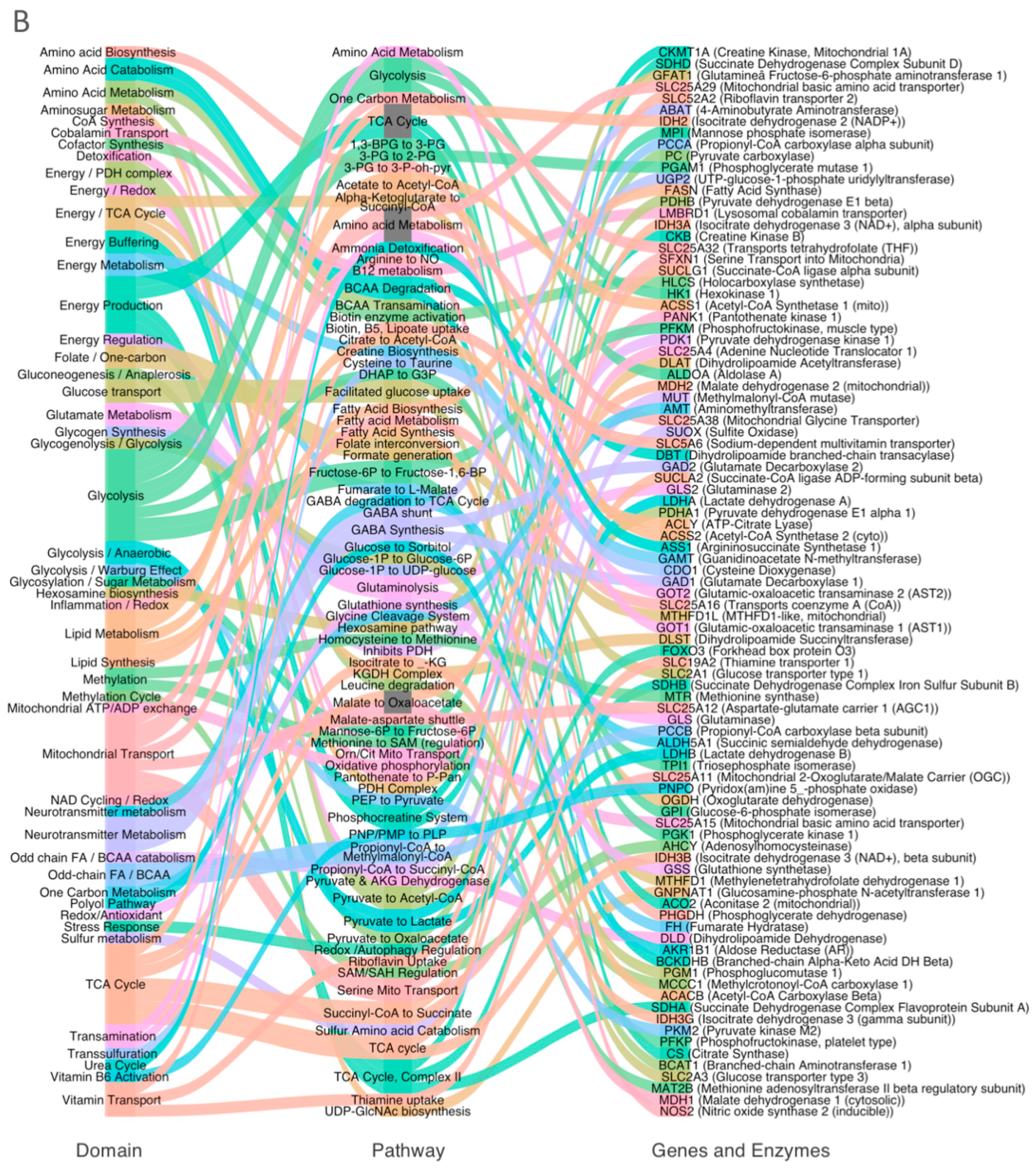
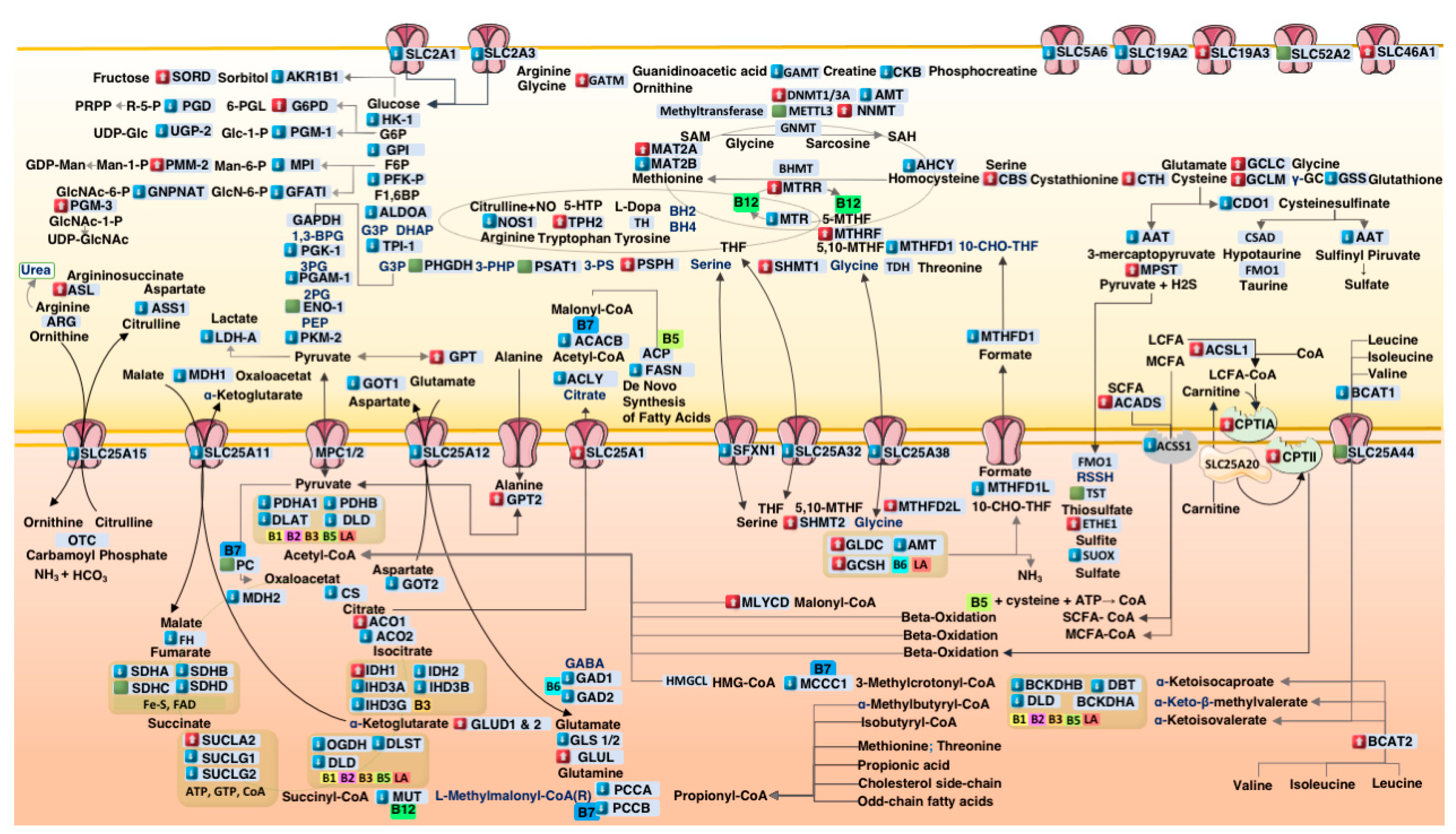
3.2. Systems-Level Visualization of Metabolic Dysregulation in ASD
3.3. Coordinated Downregulation of Cofactor Transporters and Related Enzymes
3.4. Domain-Specific Metabolic Disruption in ASD Cortex
- (A).
- Glycolysis and pentose phosphate pathway (PPP);
- (B).
- Redox, sulfur, and one-carbon metabolism;
- (C).
- Mitochondrial metabolism.
4. Discussion
4.1. Suppression of Cofactor Transporters in ASD Cortex
4.2. Glycolytic Downregulation and Citrate–Pyruvate Feedback
4.3. Mitochondrial Bottlenecks in the TCA Cycle
4.4. Propionic Acid as a Mitochondrial Toxin
4.5. Upregulation of Sulfur Metabolism Pathways in ASD
4.6. One-Carbon Metabolism Disruption in ASD
4.7. Therapeutic Implications
4.8. Study Limitations and Future Directions
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, G.; Geschwind, D.H. Genetics of autism spectrum disorder. Handb. Clin. Neurol. 2018, 147, 321–329. [Google Scholar] [PubMed]
- Rossignol, D.A.; McCarty, P.J.; Frye, R.E.; Rincon, N.; Scheck, A.C.; Brister, D. Biomarkers of mitochondrial dysfunction in autism spectrum disorder: A systematic review and meta-analysis. Neurobiol. Dis. 2024, 197, 106520. [Google Scholar]
- Frye, R.E.; Delatorre, R.; Taylor, H.; Slattery, J.; Melnyk, S.; Chowdhury, N.; James, S.J. Redox metabolism abnormalities in autistic children associated with mitochondrial disease. Transl. Psychiatry 2013, 3, e273. [Google Scholar] [CrossRef]
- Gu, F.; Chauhan, V.; Chauhan, A. Glutathione redox imbalance in brain disorders. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 89–95. [Google Scholar] [CrossRef]
- Anitha, A.; Nakamura, K.; Thanseem, I.; Matsuzaki, H.; Miyachi, T.; Tsujii, M.; Iwata, Y.; Suzuki, K.; Sugiyama, T.; Mori, N. Downregulation of the expression of mitochondrial electron transport complex genes in autism brains. Brain Pathol. 2013, 23, 294–302. [Google Scholar] [CrossRef]
- Knight, R.; McDonald, D.; Lozupone, C.; Hornig, M.; Gilbert, J.A.; Debelius, J. Towards large-cohort comparative studies to define the factors influencing the gut microbial community structure of ASD patients. Microb. Ecol. Health Dis. 2015, 26, 26555. [Google Scholar]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef]
- Naviaux, R.K. Perspective: Cell danger response Biology—The new science that connects environmental health with mitochondria and the rising tide of chronic illness. Mitochondrion 2020, 51, 40–45. [Google Scholar] [CrossRef]
- Cheng, B.; He, Y.; He, Y. Identification of Bacterial Lipopolysaccharide-Associated Genes and Molecular Subtypes in Autism Spectrum Disorder. Pharmacogenomics Pers. Med. 2025, 18, 1–18. [Google Scholar] [CrossRef]
- Lakhan, R.; Said, H.M. Lipopolysaccharide inhibits colonic biotin uptake via interference with membrane expression of its transporter: A role for a casein kinase 2-mediated pathway. Am. J. Physiol.—Cell Physiol. 2017, 312, C376–C384. [Google Scholar] [CrossRef] [PubMed]
- Anthonymuthu, S.; Sabui, S.; Lee, K.; Sheikh, A.; Fleckenstein, J.M.; Said, H.M. Bacterial lipopolysaccharide inhibits colonic carrier-mediated uptake of thiamin pyrophosphate: Roles for TLR4 receptor and NF-B/P38/JNK signaling pathway. Am. J. Physiol.—Cell Physiol. 2023, 325, C758–C769. [Google Scholar] [CrossRef] [PubMed]
- Anthonymuthu, S.; Sabui, S.; Manzon, K.I.; Sheikh, A.; Fleckenstein, J.M.; Said, H.M. Bacterial lipopolysaccharide inhibits free thiamin uptake along the intestinal tract via interference with membrane expression of thiamin transporters 1 and 2. Am. J. Physiol.—Cell Physiol. 2024, 327, C1163–C1177. [Google Scholar] [CrossRef] [PubMed]
- Irimia, M.; Weatheritt, R.J.; Ellis, J.D.; Parikshak, N.N.; Gonatopoulos-Pournatzis, T.; Babor, M.; Quesnel-Vallières, M.; Tapial, J.; Raj, B.; O’Hanlon, D.; et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 2014, 159, 1511–1523. [Google Scholar] [CrossRef]
- Kim, H.; Sriutaisuk, S.; Lee, S. Using the Tidyverse Package in R for Simulation Studies in SEM. Struct. Equ. Model. A Multidiscip. J. 2020, 27, 468–482. [Google Scholar]
- Wickham, H.; Chang, W.; Wickham, M.H. ggplot2: Create Elegant Data Visualisations Using the Grammar of Graphics, ggplot2 version 3.5.1; RStudio: Boston, MA, USA, 2016. [Google Scholar]
- Gu, Z. Complex heatmap visualization. iMeta 2022, 1, e43. [Google Scholar] [CrossRef]
- LeDell, E. Package ‘h2o’. April 2020. [Google Scholar]
- Schänzer, A.; Döring, B.; Ondrouschek, M.; Goos, S.; Garvalov, B.K.; Geyer, J.; Acker, T.; Neubauer, B.; Hahn, A. Stress-induced upregulation of SLC19A3 is impaired in biotin-thiamine- responsive basal ganglia disease. Brain Pathol. 2014, 24, 270–279. [Google Scholar] [CrossRef]
- Parker, S.J.; Amendola, C.R.; Hollinshead, K.E.; Yu, Q.; Yamamoto, K.; Encarnación-Rosado, J.; Rose, R.E.; LaRue, M.M.; Sohn, A.S.; Biancur, D.E.; et al. Selective alanine transporter utilization creates a targetable metabolic niche in pancreatic cancer. Cancer Discov. 2020, 10, 1018–1037. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef]
- Giulivi, C.; Zhang, Y.F.; Omanska-Klusek, A.; Ross-Inta, C.; Wong, S.; Hertz-Picciotto, I.; Tassone, F.; Pessah, I.N. Mitochondrial Dysfunction in Autism. JAMA 2010, 304, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K. Metabolic features of the cell danger response. Mitochondrion 2014, 16, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Macfabe, D.F. Short-chain fatty acid fermentation products of the gut microbiome: Implications in autism spectrum disorders. Microb. Ecol. Health Dis. 2012, 23, 19260. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Rose, S.; Chacko, J.; Wynne, R.; Bennuri, S.C.; Slattery, J.C.; Tippett, M.; Delhey, L.; Melnyk, S.; Kahler, S.G.; et al. Modulation of mitochondrial function by the microbiome metabolite propionic acid in autism and control cell lines. Transl. Psychiatry 2016, 6, e927. [Google Scholar] [CrossRef]
- SMurch, S.H.; MacDonald, T.T.; Walker-Smith, J.A.; Lionetti, P.; Levin, M.; Klein, N.J. Disruption of sulphated glycosaminoglycans in intestinal inflammation. Lancet 1993, 341, 711–714. [Google Scholar] [CrossRef]
- Kane, R.E.; Li, A.P.; Kaminski, D.R. Sulfation and glucuronidation of acetaminophen by human hepatocytes cultured on Matrigel and type 1 collagen reproduces conjugation in vivo. Drug Metab. Dispos. 1995, 23, 303–307. [Google Scholar] [CrossRef]
- Sun, Y.; Harps, L.C.; Bureik, M.; Parr, M.K. Human Sulfotransferase Assays With PAPS Production in situ. Front. Mol. Biosci. 2022, 9, 827638. [Google Scholar] [CrossRef]
- Tiranti, V.; Viscomi, C.; Hildebrandt, T.; Di Meo, I.; Mineri, R.; Tiveron, C.; DLevitt, M.; Prelle, A.; Fagiolari, G.; Rimoldi, M.; et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 2009, 15, 200–205. [Google Scholar] [CrossRef]
- Waring, R.H.; Klovrza, L.V. Sulphur metabolism in autism. J. Nutr. Environ. Med. 2000, 10, 25–32. [Google Scholar] [CrossRef]
- Garrett, R.M.; Johnson, J.L.; Graf, T.N.; Feigenbaum, A.; Rajagopalan, K.V. Human sulfite oxidase R160Q: Identification of the mutation in a sulfite oxidase-deficient patient and expression and characterization of the mutant enzyme. Proc. Natl. Acad. Sci. USA 1998, 95, 6394–6398. [Google Scholar] [CrossRef]
- Grings, M.; Moura, A.P.; Amaral, A.U.; Parmeggiani, B.; Gasparotto, J.; Moreira, J.C.; Gelain, D.P.; Wyse, A.T.; Wajner, M.; Leipnitz, G. Sulfite disrupts brain mitochondrial energy homeostasis and induces mitochondrial permeability transition pore opening via thiol group modification. Biochim. Biophys. Acta—Mol. Basis Dis. 2014, 1842, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.; Duran, M.; Poll-The, B.-T.; Surtees, R.; Snell, K.; de Koning, T.J. L-serine in disease and development. Biochem. J. 2003, 371, 653–661. [Google Scholar]
- Yang, J.H.; Wada, A.; Yoshida, K.; Miyoshi, Y.; Sayano, T.; Esaki, K.; Kinoshita, M.O.; Tomonaga, S.; Azuma, N.; Watanabe, M.; et al. Brain-specific Phgdh deletion reveals a pivotal role for l-serine biosynthesis in controlling the level of D-serine, an N-methyl-D-aspartate receptor co-agonist, in adult brain. J. Biol. Chem. 2010, 285, 41380–41390. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | %Δ Expression | log2FC | AUC | Cofactor(s) | Pathway | Functional Role |
|---|---|---|---|---|---|---|---|
| SUCLA2 | Succinate-CoA ligase (ADP-forming β) | −11.93 | −0.180 | 0.88 | ADP, CoA | TCA Cycle | Succinyl–CoA to Succinate |
| GOT1 | Aspartate transaminase 1 (AST1) | −11.82 | −0.181 | 0.86 | PLP (Vitamin B6) | Transamination | Glutamate/Oxaloacetate Shuttle |
| SLC25A11 | 2-Oxoglutarate/malate carrier (OGC) | −7.99 | −0.120 | 0.85 | Mitochondrial Transport | Malate Shuttle (Mito) | |
| CKMT1A | Mitochondrial creatine kinase 1A | −16.9 | −0.267 | 0.84 | Mg2+, ATP | Energy Buffering | Phosphocreatine Shuttle |
| GPT2 | Alanine transaminase 2 (ALT2) | 4.64 | 0.065 | 0.83 | PLP (Vitamin B6) | Transamination | Alanine ↔ Pyruvate |
| GOT2 | Aspartate transaminase 2 (AST2) | −8.48 | −0.128 | 0.80 | PLP (Vitamin B6) | Transamination | Mitochondrial Transamination |
| GLS | Glutaminase | −8.08 | −0.122 | 0.79 | Glutamine | Glutamate Metabolism | Glutamine to Glutamate |
| IDH3A | Isocitrate dehydrogenase 3α(NAD+-linked) | −8.33 | −0.125 | 0.79 | NAD+ | TCA Cycle | Isocitrate to α-Ketoglutarate |
| MDH2 | Malate dehydrogenase 2 (mitochondrial) | −5.93 | −0.088 | 0.78 | NAD+ | TCA Cycle | Malate to Oxaloacetate (mito) |
| IDH3B | Isocitrate dehydrogenase3β (NAD+-linked) | −8.13 | −0.122 | 0.78 | NAD+ | TCA Cycle | Isocitrate to α-Ketoglutarate |
| SHMT1 | Serine hydroxymethyltransferase 1 | 7.55 | 0.105 | 0.77 | PLP (Vitamin B6) | One Carbon Metabolism | Serine to Glycine |
| MDH1 | Malate dehydrogenase 1 (cytosolic) | −11.78 | −0.181 | 0.74 | NAD+ | NAD Cycling/Redox | Malate to Oxaloacetate |
| OGDH | Oxoglutarate dehydrogenase | −3.63 | −0.053 | 0.73 | B1, NAD+, CoA, lipoate | Energy/TCA Cycle | α-Ketoglutarate to Succinyl-CoA |
| PDHA1 | Pyruvate dehydrogenase E1 α subunit | −2.25 | −0.033 | 0.73 | B1, NAD+, CoA, lipoate | Energy/PDH complex | Pyruvate to Acetyl-CoA |
| MAT2B | Methionine adenosyltransferase II β | −11.9 | −0.184 | 0.72 | Methylation | Methionine to SAM (Regulation) | |
| SFXN1 | Serine transport into mitochondria | −5.51 | −0.082 | 0.71 | Mitochondrial Transport | Mitochondrial Serine Transport | |
| MTHFD1 | Methylene-THF dehydrogenase 1 | −4.73 | −0.070 | 0.71 | NADH+ | Folate/One-Carbon Metabolism | Folate Interconversion |
| DLD | Dihydrolipoamide dehydrogenase | −9.0 | −0.136 | 0.71 | FAD, NAD | Energy/Redox | E3 Subunit of PDH/OGDH Complexes |
| SUOX | Sulfite oxidase | −6.6 | −0.097 | 0.70 | Molybdenum, Cytochrome b5, and Heme | Sulfur Metabolism | Sulfite Oxidation |
| TST | Thiosulfate sulfurtransferase (rhodanese) | 0.36 | 0.005 | 0.70 | Detoxification | Thiosulfate Sulfurtransferase (Detoxification) | |
| SUCLG2 | Succinate-CoA ligase (GDP-forming β) | 13.83 | 0.187 | 0.69 | GDP, CoA | TCA Cycle | Succinyl-CoA to Succinate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dervishi, A. A Systems Hypothesis of Lipopolysaccharide-Induced Vitamin Transport Suppression and Metabolic Reprogramming in Autism Spectrum Disorders: An Open Call for Validation and Therapeutic Translation. Metabolites 2025, 15, 399. https://doi.org/10.3390/metabo15060399
Dervishi A. A Systems Hypothesis of Lipopolysaccharide-Induced Vitamin Transport Suppression and Metabolic Reprogramming in Autism Spectrum Disorders: An Open Call for Validation and Therapeutic Translation. Metabolites. 2025; 15(6):399. https://doi.org/10.3390/metabo15060399
Chicago/Turabian StyleDervishi, Albion. 2025. "A Systems Hypothesis of Lipopolysaccharide-Induced Vitamin Transport Suppression and Metabolic Reprogramming in Autism Spectrum Disorders: An Open Call for Validation and Therapeutic Translation" Metabolites 15, no. 6: 399. https://doi.org/10.3390/metabo15060399
APA StyleDervishi, A. (2025). A Systems Hypothesis of Lipopolysaccharide-Induced Vitamin Transport Suppression and Metabolic Reprogramming in Autism Spectrum Disorders: An Open Call for Validation and Therapeutic Translation. Metabolites, 15(6), 399. https://doi.org/10.3390/metabo15060399