
Comparison of Pyrazinamide with Isoniazid for Their Effects on the Heme Biosynthetic Pathway in Mouse Liver

 , and
, and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animals and Treatment
2.3. Quantitative Polymerase Chain Reaction (qPCR) Analysis
2.4. Western Blotting Analysis
2.5. ALA Analysis
2.6. PPIX Analysis
2.7. UPLC-QTOFMS Analysis
2.8. Metabolomic Analysis
2.9. Assessment of Liver Injury
2.10. Statistical Analysis
3. Results
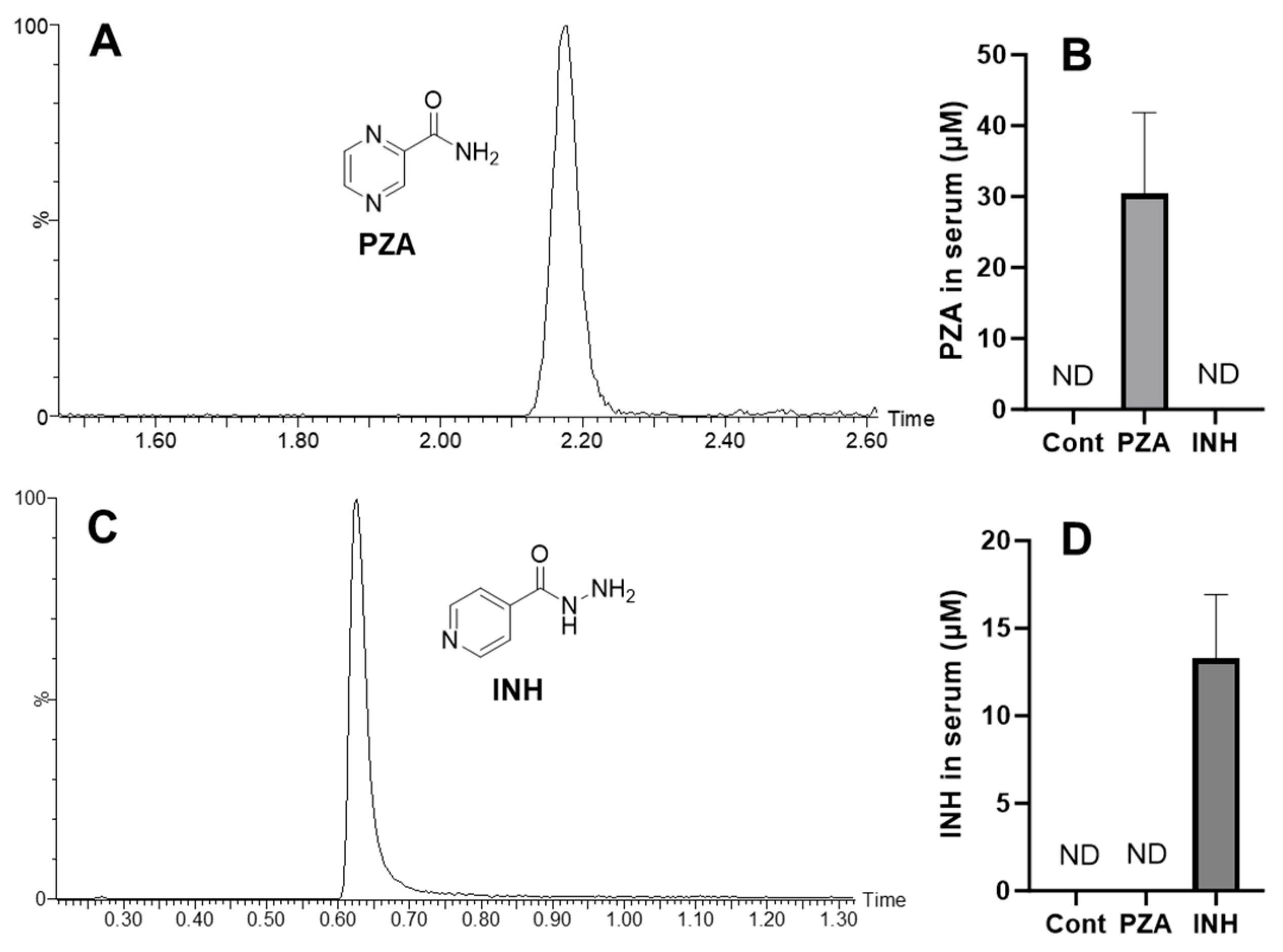
3.1. The Exposure of PZA and INH to Mice
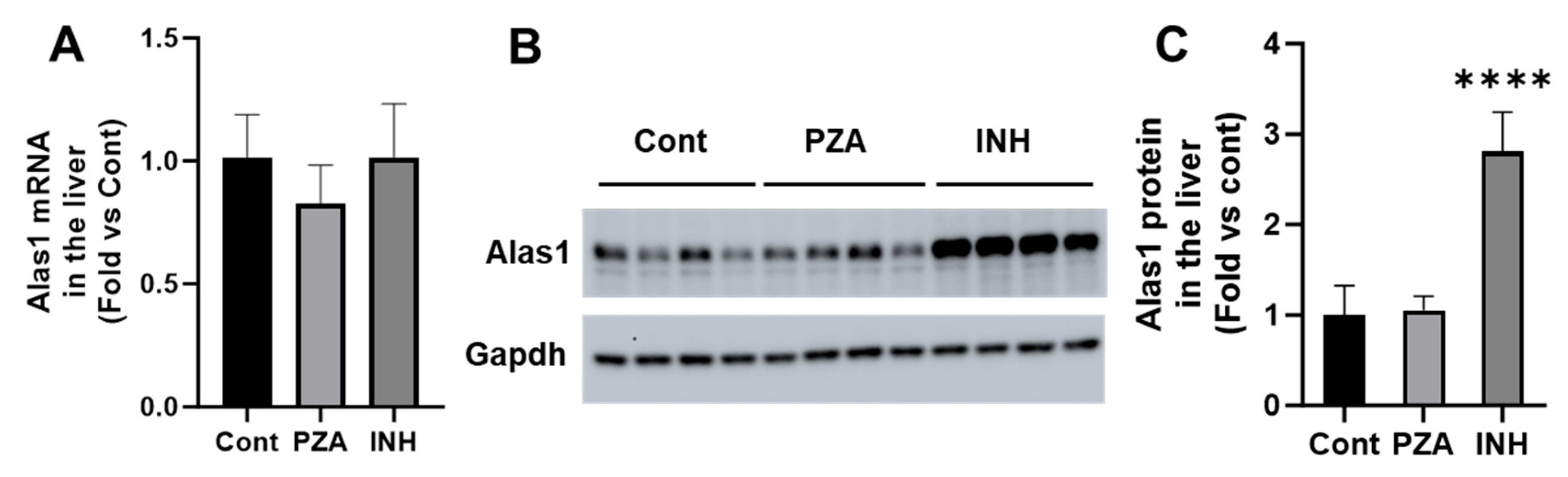
3.2. Effects of PZA and INH on Alas1 Expression in the Liver
3.3. Effects of PZA and INH on ALA Levels in the Liver and Urine
3.4. Effects of PZA and INH on Fech Expression in the Liver
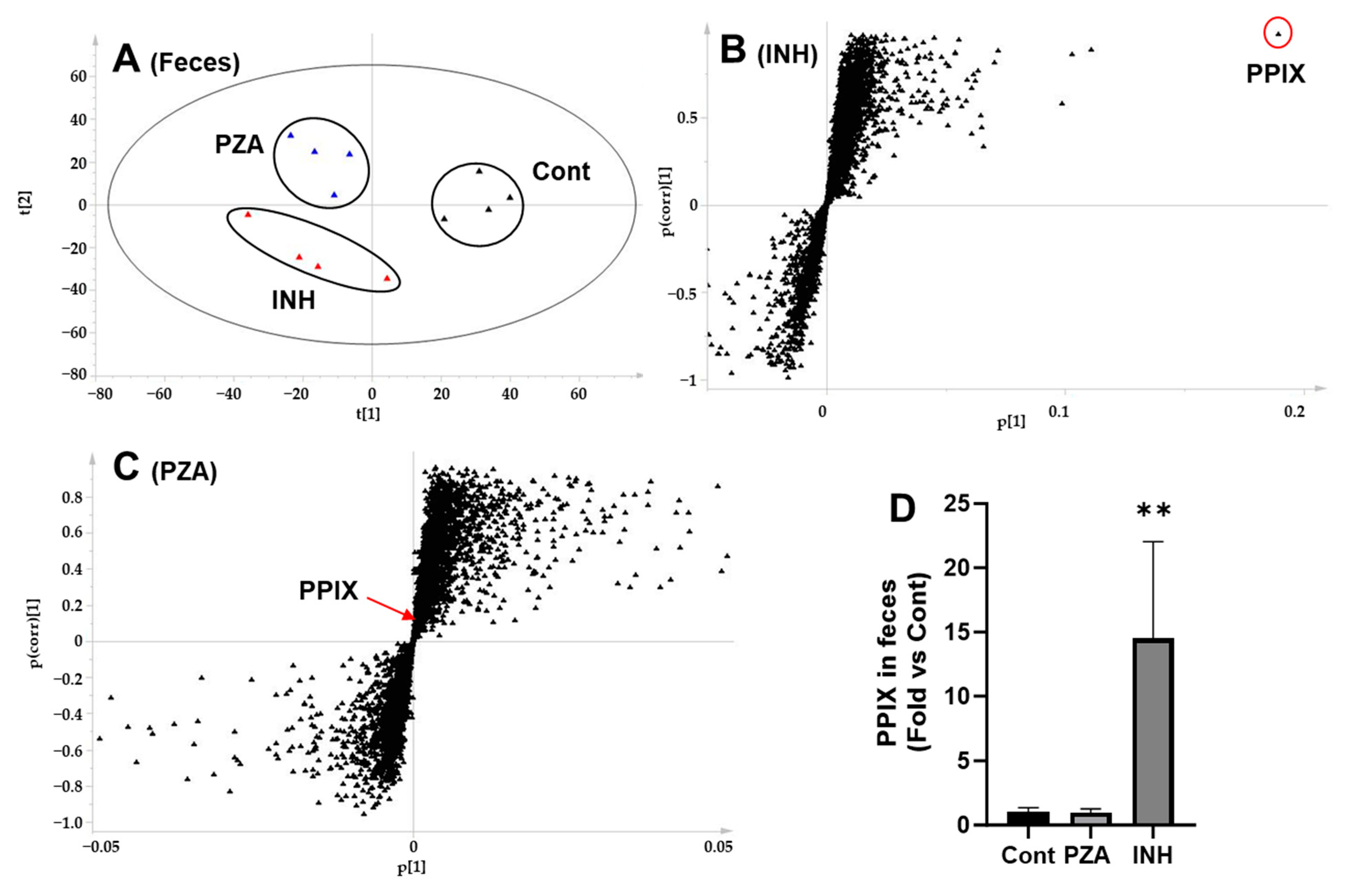
3.5. Effects of PZA and INH on PPIX Levels in the Liver and Feces
3.6. Effects of PZA and INH on Liver Function
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALA | Aminolevulinic acid |
| ALAS | Aminolevulinic acid synthase |
| ALT | Alanine transaminase |
| ALP | Alkaline phosphatase |
| AQC | 6-Aminoquinolyl-N-hydroxysuccinimidyl carbamate |
| AST | Aspartate transaminase |
| FECH | Ferrochelatase |
| GADPH | Glyceraldehyde 3-phosphate dehydrogenase |
| INH | Isoniazid |
| 15N,13C5-ALA | 15N,13C5-Aminolevulinic acid |
| OPLS-DA | Orthogonal partial least-squares discriminant analysis |
| PA | Pyrazinoic acid |
| PCA | Principal component analysis |
| PPIX | Protoporphyrin IX |
| PZA | Pyrazinamide |
| QTOFMS | Quadrupole time of flight mass spectrometry |
| UPLC | Ultra-performance liquid chromatography |
| TB | Tuberculosis |
References
- WHO. Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed on 1 May 2025).
- Cade, C.E.; Dlouhy, A.C.; Medzihradszky, K.F.; Salas-Castillo, S.P.; Ghiladi, R.A. Isoniazid-resistance conferring mutations in Mycobacterium tuberculosis KatG: Catalase, peroxidase, and INH-NADH adduct formation activities. Protein Sci. 2010, 19, 458–474. [Google Scholar] [CrossRef] [PubMed]
- Rawat, R.; Whitty, A.; Tonge, P.J. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: Adduct affinity and drug resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 13881–13886. [Google Scholar] [CrossRef]
- Nono, V.N.; Nantia, E.A.; Mutshembele, A.; Teagho, S.N.; Simo, Y.W.K.; Takong, B.S.; Djieugoue, Y.J.; Assolo, Y.P.; Ongboulal, S.M.; Awungafac, S.N.; et al. Prevalence of katG and inhA mutations associated with isoniazid resistance in Mycobacterium tuberculosis clinical isolates in Cameroon. BMC Microbiol. 2025, 25, 127. [Google Scholar] [CrossRef] [PubMed]
- Scorpio, A.; Zhang, Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 1996, 2, 662–667. [Google Scholar] [CrossRef]
- Cheng, S.J.; Thibert, L.; Sanchez, T.; Heifets, L.; Zhang, Y. pncA mutations as a major mechanism of pyrazinamide resistance in Mycobacterium tuberculosis: Spread of a monoresistant strain in Quebec, Canada. Antimicrob. Agents Chemother. 2000, 44, 528–532. [Google Scholar] [CrossRef]
- Gopal, P.; Sarathy, J.P.; Yee, M.; Ragunathan, P.; Shin, J.; Bhushan, S.; Zhu, J.; Akopian, T.; Kandror, O.; Lim, T.K.; et al. Pyrazinamide triggers degradation of its target aspartate decarboxylase. Nat. Commun. 2020, 11, 1661. [Google Scholar] [CrossRef]
- Oluoch, P.O.; Koh, E.I.; Proulx, M.K.; Reames, C.J.; Papavinasasundaram, K.G.; Murphy, K.C.; Zimmerman, M.D.; Dartois, V.; Sassetti, C.M. Chemical genetic interactions elucidate pathways controlling tuberculosis antibiotic efficacy during infection. Proc. Natl. Acad. Sci. USA 2025, 122, e2417525122. [Google Scholar] [CrossRef] [PubMed]
- Khanderia, U.; Bhattacharya, A. Acute intermittent porphyria: Pathophysiology and treatment. Pharmacotherapy 1984, 4, 144–150. [Google Scholar] [CrossRef]
- Boon, F.F.; Ellis, C. Acute intermittent porphyria in a children’s psychiatric hospital. J. Am. Acad. Child Adolesc. Psychiatry 1989, 28, 606–609. [Google Scholar] [CrossRef]
- Santosh, P.J.; Malhotra, S. Varied psychiatric manifestations of acute intermittent porphyria. Biol. Psychiatry 1994, 36, 744–747. [Google Scholar] [CrossRef]
- Anderson, K.E.; Bloomer, J.R.; Bonkovsky, H.L.; Kushner, J.P.; Pierach, C.A.; Pimstone, N.R.; Desnick, R.J. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann. Intern. Med. 2005, 142, 439–450. [Google Scholar] [CrossRef]
- Spiritos, Z.; Salvador, S.; Mosquera, D.; Wilder, J. Acute Intermittent Porphyria: Current Perspectives And Case Presentation. Ther. Clin. Risk Manag. 2019, 15, 1443–1451. [Google Scholar] [CrossRef]
- Balwani, M.; Desnick, R.J. The porphyrias: Advances in diagnosis and treatment. Blood 2012, 120, 4496–4504. [Google Scholar] [CrossRef]
- Phillips, J.D. Heme biosynthesis and the porphyrias. Mol. Genet. Metab. 2019, 128, 164–177. [Google Scholar] [CrossRef]
- Taylor, J.L.; Brown, B.L. Structural basis for dysregulation of aminolevulinic acid synthase in human disease. J. Biol. Chem. 2022, 298, 101643. [Google Scholar] [CrossRef]
- Medlock, A.; Swartz, L.; Dailey, T.A.; Dailey, H.A.; Lanzilotta, W.N. Substrate interactions with human ferrochelatase. Proc. Natl. Acad. Sci. USA 2007, 104, 1789–1793. [Google Scholar] [CrossRef]
- Sachar, M.; Li, F.; Liu, K.; Wang, P.; Lu, J.; Ma, X. Chronic Treatment with Isoniazid Causes Protoporphyrin IX Accumulation in Mouse Liver. Chem. Res. Toxicol. 2016, 29, 1293–1297. [Google Scholar] [CrossRef]
- Brewer, C.T.; Yang, L.; Edwards, A.; Lu, Y.; Low, J.; Wu, J.; Lee, R.E.; Chen, T. The Isoniazid Metabolites Hydrazine and Pyridoxal Isonicotinoyl Hydrazone Modulate Heme Biosynthesis. Toxicol. Sci. 2019, 168, 209–224. [Google Scholar] [CrossRef]
- Granick, S. The induction in vitro of the synthesis of delta-aminolevulinic acid synthetase in chemical porphyria: A response to certain drugs, sex hormones, and foreign chemicals. J. Biol. Chem. 1966, 241, 1359–1375. [Google Scholar] [CrossRef]
- Treece, G.L.; Magnussen, C.R.; Patterson, J.R.; Tschudy, D.P. Exacerbation of porphyria during treatment of pulmonary tuberculosis. Am. Rev. Respir. Dis. 1976, 113, 233–237. [Google Scholar]
- Hussain, Z.; Qi, Q.; Zhu, J.; Anderson, K.E.; Ma, X. Protoporphyrin IX-induced phototoxicity: Mechanisms and therapeutics. Pharmacol. Ther. 2023, 248, 108487. [Google Scholar] [CrossRef]
- Kuo, H.C.; Ro, L.S.; Lin, C.N.; Chu, C.C.; Liao, M.F.; Chang, H.S. Baseline urinary ALA and PBG as criteria for starting pharmacologic prophylactic treatment in acute intermittent porphyria treated with givosiran. Mol. Genet. Metab. Rep. 2024, 41, 101169. [Google Scholar] [CrossRef]
- Minder, A.E.; Kluijver, L.G.; Barman-Aksozen, J.; Minder, E.I.; Langendonk, J.G. Erythropoietic protoporphyrias: Pathogenesis, diagnosis and management. Liver Int. 2025, 45, e16027. [Google Scholar] [CrossRef]
- Wang, P.; Sachar, M.; Lu, J.; Shehu, A.I.; Zhu, J.; Chen, J.; Liu, K.; Anderson, K.E.; Xie, W.; Gonzalez, F.J.; et al. The essential role of the transporter ABCG2 in the pathophysiology of erythropoietic protoporphyria. Sci. Adv. 2019, 5, eaaw6127. [Google Scholar] [CrossRef] [PubMed]
- Dickey, A.K.; Leaf, R.K.; Balwani, M. Update on the Porphyrias. Annu. Rev. Med. 2024, 75, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Siegesmund, M.; van Tuyll van Serooskerken, A.M.; Poblete-Gutierrez, P.; Frank, J. The acute hepatic porphyrias: Current status and future challenges. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 593–605. [Google Scholar] [CrossRef]
- Gonzalez, F.J. The 2006 Bernard B. Brodie Award Lecture. Cyp2e1. Drug Metab. Dispos. 2007, 35, 1–8. [Google Scholar] [CrossRef]
- Park, K.S.; Sohn, D.H.; Veech, R.L.; Song, B.J. Translational activation of ethanol-inducible cytochrome P450 (CYP2E1) by isoniazid. Eur. J. Pharmacol. 1993, 248, 7–14. [Google Scholar] [CrossRef]
- Roberts, B.J.; Song, B.J.; Soh, Y.; Park, S.S.; Shoaf, S.E. Ethanol induces CYP2E1 by protein stabilization. Role of ubiquitin conjugation in the rapid degradation of CYP2E1. J. Biol. Chem. 1995, 270, 29632–29635. [Google Scholar] [CrossRef] [PubMed]
- Song, B.J.; Matsunaga, T.; Hardwick, J.P.; Park, S.S.; Veech, R.L.; Yang, C.S.; Gelboin, H.V.; Gonzalez, F.J. Stabilization of cytochrome P450j messenger ribonucleic acid in the diabetic rat. Mol. Endocrinol. 1987, 1, 542–547. [Google Scholar] [CrossRef]
- Sotgiu, G.; Centis, R.; D’Ambrosio, L.; Migliori, G.B. Tuberculosis treatment and drug regimens. Cold Spring Harb. Perspect. Med. 2015, 5, a017822. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, F.-Y.; Gu, R.; Zhang, J.; Weiss, J.L.; Lu, J.; Ma, Q.; Ma, X. Comparison of Pyrazinamide with Isoniazid for Their Effects on the Heme Biosynthetic Pathway in Mouse Liver. Metabolites 2025, 15, 355. https://doi.org/10.3390/metabo15060355
Qin F-Y, Gu R, Zhang J, Weiss JL, Lu J, Ma Q, Ma X. Comparison of Pyrazinamide with Isoniazid for Their Effects on the Heme Biosynthetic Pathway in Mouse Liver. Metabolites. 2025; 15(6):355. https://doi.org/10.3390/metabo15060355
Chicago/Turabian StyleQin, Fu-Ying, Ruizhi Gu, Jiaojiao Zhang, Jaden Leigh Weiss, Jie Lu, Qing Ma, and Xiaochao Ma. 2025. "Comparison of Pyrazinamide with Isoniazid for Their Effects on the Heme Biosynthetic Pathway in Mouse Liver" Metabolites 15, no. 6: 355. https://doi.org/10.3390/metabo15060355
APA StyleQin, F.-Y., Gu, R., Zhang, J., Weiss, J. L., Lu, J., Ma, Q., & Ma, X. (2025). Comparison of Pyrazinamide with Isoniazid for Their Effects on the Heme Biosynthetic Pathway in Mouse Liver. Metabolites, 15(6), 355. https://doi.org/10.3390/metabo15060355