
Cell-Free DNA (cfDNA) Regulates Metabolic Remodeling in the ES-2 Ovarian Carcinoma Cell Line, Influencing Cell Proliferation, Quiescence, and Chemoresistance in a Cell-of-Origin-Specific Manner

 , , ,
, , ,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Genomic Material Extraction
2.3. Cell Proliferation Assessment
2.4. Wound Healing Assay
2.5. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.6. Cell Death Analysis
2.7. Immunofluorescence Analysis
2.8. Quantification of Cytoplasmic ROS by Flow Cytometry
2.9. Quantification of Lipid Peroxides by Flow Cytometry
2.10. Lentiviral Transduction
2.11. Co-Culture of ES-2 and ES-2-GFP Cells
2.12. Analysis of Ki67 Marker by Flow Cytometry
2.13. Quantitative Real-Time PCR (qPCR)
2.14. Generation of Spheroids
2.15. Spheroid Sprouting
2.16. Statistical Analysis
3. Results
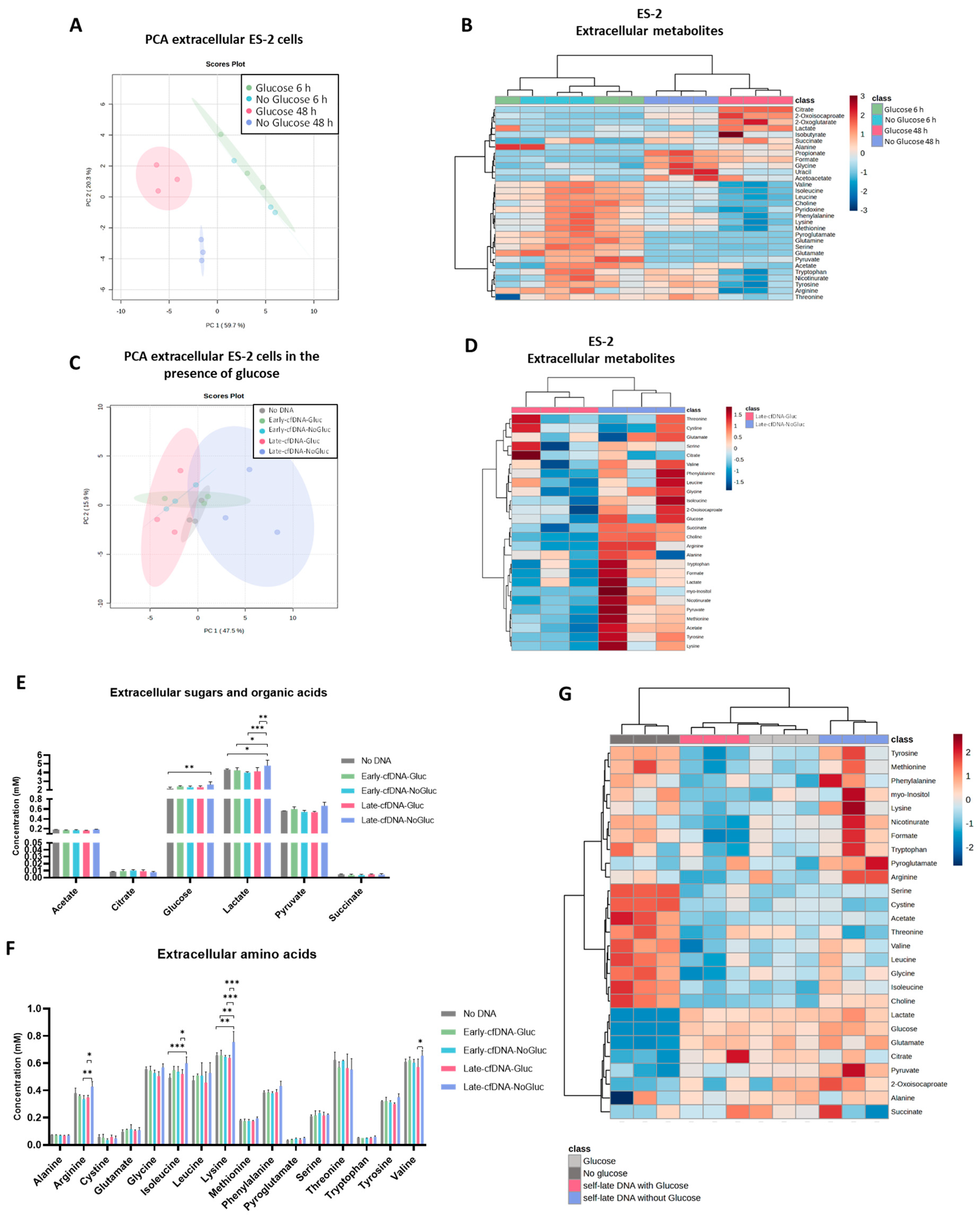
3.1. Glucose Availability Impacts the Exometabolome of Cell Subsets Cultured During 48 H
3.2. The Late cfDNAs Promote a Metabolic Remodeling in Unselected ES-2 Cells, Resembling the Metabolic Profile of the Cells of Origin of These cfDNAs
3.3. The Metabolic Remodeling Promoted by Late-cfDNA-Gluc Sustains Cell Proliferation of Unselected Cells, but upon Long-Term Selection, Late-cfDNA-Gluc Induced Quiescence
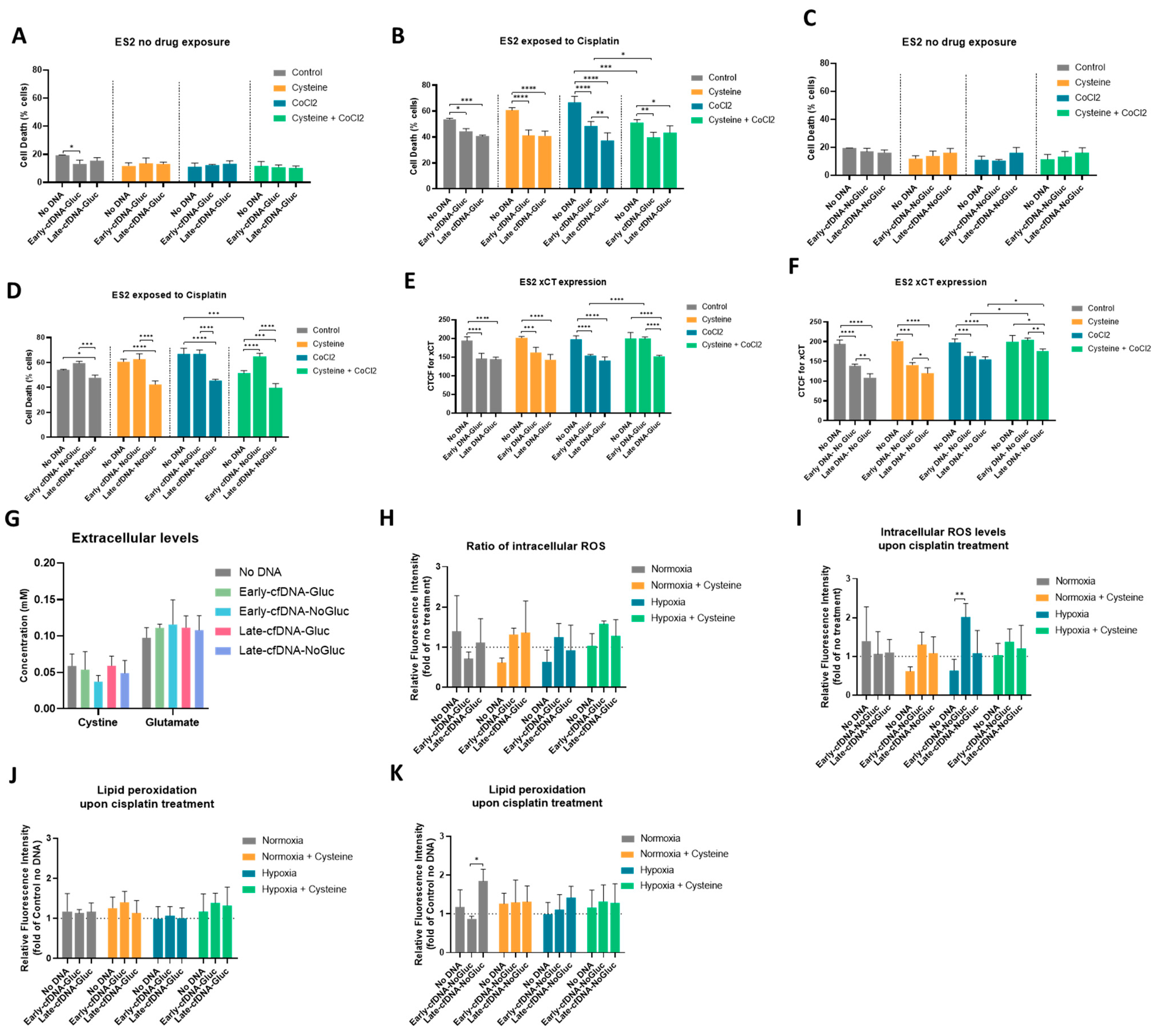
3.4. All cfDNA Variants Promoted Cisplatin Resistance in ES-2 Cells, and Cysteine Protects Cells from Cisplatin in Hypoxia-Mimicked Conditions, Potentiating the Early-cfDNA-Gluc Protection
3.5. xCT Cyst(e)ine Antiporter May Be Involved in the Concomitant Protective Effect Between Cysteine and cfDNA
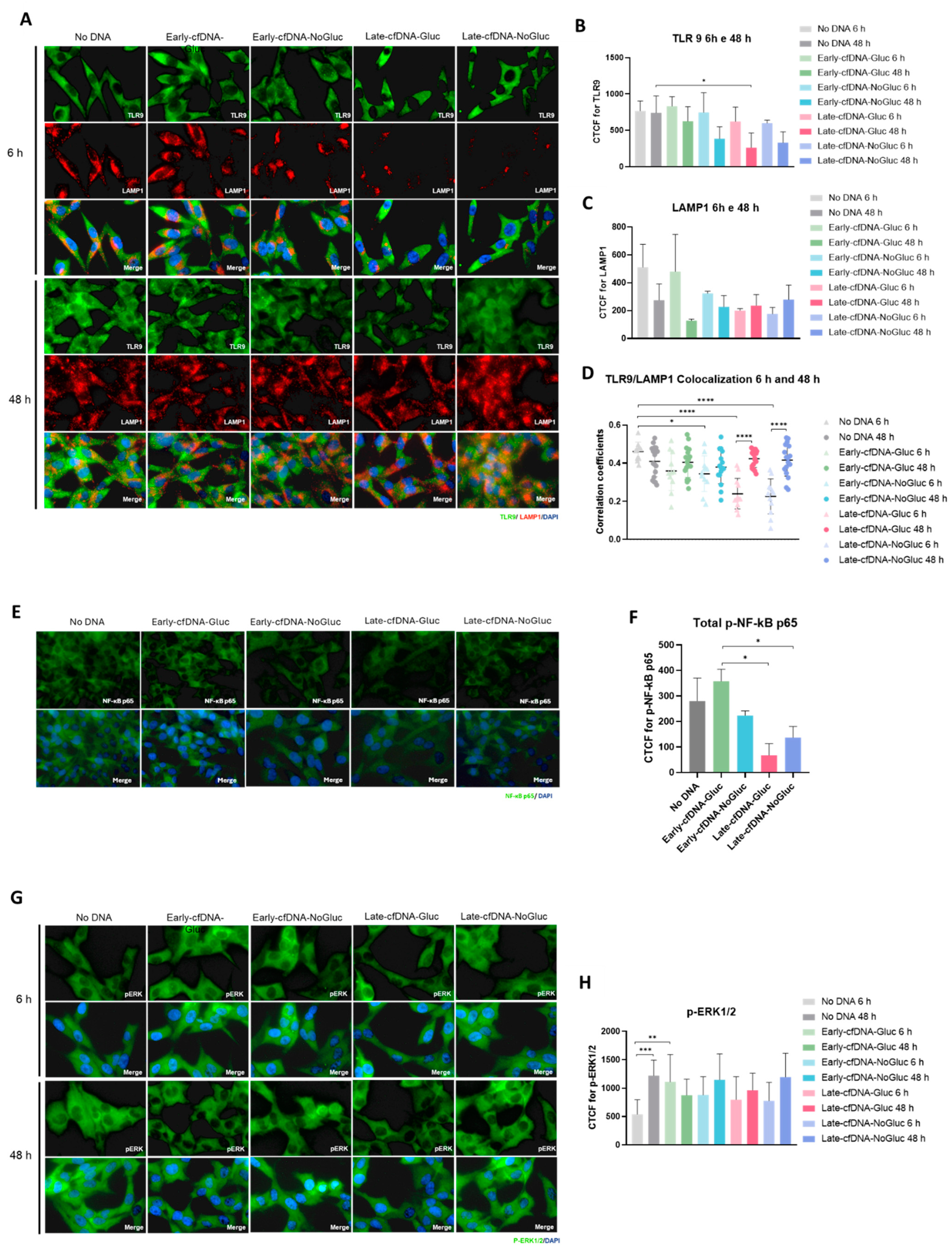
3.6. All cfDNA Variants Decreased Toll-like Receptor 9 (TLR9) Levels
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pessoa, L.S.; Heringer, M.; Ferrer, V.P. ctDNA as a cancer biomarker: A broad overview. Crit. Rev. Oncol. Hematol. 2020, 155, 103109. [Google Scholar] [CrossRef] [PubMed]
- Telekes, A.; Horváth, A. The Role of Cell-Free DNA in Cancer Treatment Decision Making. Cancers 2022, 14, 6115. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Haglund, T.A.; Rogers, A.J.; Ghanim, A.F.; Sethu, P. Review: Microfluidics technologies for blood-based cancer liquid biopsies. Anal. Chim. Acta 2018, 1012, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Mandel, P.; Metais, P. Nuclear Acids In Human Blood Plasma. C R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar] [PubMed]
- Stroun, M.; Maurice, P.; Vasioukhin, V.; Lyautey, J.; Lederrey, C.; Lefort, F.; Rossier, A.; Chen, X.Q.; Anker, P. The origin and mechanism of circulating DNA. Ann. N. Y. Acad. Sci. 2000, 906, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, M.; Baranova, A.; Butler, T.; Spellman, P.; Mileyko, V. Non-random fragmentation patterns in circulating cell-free DNA reflect epigenetic regulation. BMC Genom. 2015, 16 (Suppl. S13), S1. [Google Scholar] [CrossRef]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.N.; Swaminathan, R. Overview of circulating nucleic acids in plasma/serum. Ann. N. Y. Acad. Sci. 2008, 1137, 236–242. [Google Scholar] [CrossRef]
- Volik, S.; Alcaide, M.; Morin, R.D.; Collins, C. Cell-free DNA (cfDNA): Clinical Significance and Utility in Cancer Shaped By Emerging Technologies. Mol. Cancer Res. 2016, 14, 898–908. [Google Scholar] [CrossRef]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar]
- Yan, Y.Y.; Guo, Q.R.; Wang, F.H.; Adhikari, R.; Zhu, Z.Y.; Zhang, H.Y.; Zhou, W.M.; Yu, H.; Li, J.Q.; Zhang, J.Y. Cell-Free DNA: Hope and Potential Application in Cancer. Front. Cell Dev. Biol. 2021, 9, 639233. [Google Scholar] [CrossRef]
- Heitzer, E.; Auinger, L.; Speicher, M.R. Cell-free DNA and apoptosis: How dead cells inform about the living. Trends Mol. Med. 2020, 26, 519–528. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef]
- Muinelo-Romay, L.; Casas-Arozamena, C.; Abal, M. Liquid Biopsy in Endometrial Cancer: New Opportunities for Personalized Oncology. Int. J. Mol. Sci. 2018, 19, 2311. [Google Scholar] [CrossRef] [PubMed]
- Khakoo, S.; Georgiou, A.; Gerlinger, M.; Cunningham, D.; Starling, N. Circulating tumour DNA, a promising biomarker for the management of colorectal cancer. Crit. Rev. Oncol. Hematol. 2018, 122, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112.e114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liang, Y.; Li, S.; Zeng, F.; Meng, Y.; Chen, Z.; Liu, S.; Tao, Y.; Yu, F. The interplay of circulating tumor DNA and chromatin modification, therapeutic resistance, and metastasis. Mol. Cancer 2019, 18, 36. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Pius, C.; Nabeel, M.; Nair, M.; Vishwanatha, J.K.; Ahmad, S.; Basha, R. Ovarian cancer: Current status and strategies for improving therapeutic outcomes. Cancer Med. 2019, 8, 7018–7031. [Google Scholar]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Bowtell, D.D. The genesis and evolution of high-grade serous ovarian cancer. Nat. Rev. Cancer 2010, 10, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C., Jr.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef]
- Desai, A.; Xu, J.; Aysola, K.; Qin, Y.; Okoli, C.; Hariprasad, R.; Chinemerem, U.; Gates, C.; Reddy, A.; Danner, O.; et al. Epithelial ovarian cancer: An overview. World J. Transl. Med. 2014, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Prat, J. Ovarian carcinomas: Five distinct diseases with different origins, genetic alterations, and clinicopathological features. Virchows Arch. 2012, 460, 237–249. [Google Scholar] [CrossRef]
- Alatise, K.L.; Gardner, S.; Alexander-Bryant, A. Mechanisms of Drug Resistance in Ovarian Cancer and Associated Gene Targets. Cancers 2022, 14, 6246. [Google Scholar] [CrossRef] [PubMed]
- Gadducci, A.; Multinu, F.; Cosio, S.; Carinelli, S.; Ghioni, M.; Aletti, G.D. Clear cell carcinoma of the ovary: Epidemiology, pathological and biological features, treatment options and clinical outcomes. Gynecol. Oncol. 2021, 162, 741–750. [Google Scholar] [CrossRef]
- del Carmen, M.G.; Birrer, M.; Schorge, J.O. Clear cell carcinoma of the ovary: A review of the literature. Gynecol. Oncol. 2012, 126, 481–490. [Google Scholar] [CrossRef]
- Lopes-Coelho, F.; Gouveia-Fernandes, S.; Gonçalves, L.G.; Nunes, C.; Faustino, I.; Silva, F.; Félix, A.; Pereira, S.A.; Serpa, J. HNF1β drives glutathione (GSH) synthesis underlying intrinsic carboplatin resistance of ovarian clear cell carcinoma (OCCC). Tumour. Biol. 2016, 37, 4813–4829. [Google Scholar] [CrossRef]
- Werner, B.; Yuwono, N.; Duggan, J.; Liu, D.; David, C.; Srirangan, S.; Provan, P.; DeFazio, A.; Arora, V.; Farrell, R.; et al. Cell-free DNA is abundant in ascites and represents a liquid biopsy of ovarian cancer. Gynecol. Oncol. 2021, 162, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Werner, B.; Powell, E.; Duggan, J.; Cortesi, M.; Lee, Y.C.; Arora, V.; Athavale, R.; Dean, M.; Warton, K.; Ford, C.E. Cell-free DNA from ascites identifies clinically relevant variants and tumour evolution in patients with advanced ovarian cancer. Mol. Oncol. 2024, 18, 2668–2683. [Google Scholar] [CrossRef]
- Han, M.R.; Lee, S.H.; Park, J.Y.; Hong, H.; Ho, J.Y.; Hur, S.Y.; Choi, Y.J. Clinical Implications of Circulating Tumor DNA from Ascites and Serial Plasma in Ovarian Cancer. Cancer Res. Treat. 2020, 52, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, X.; Liu, Q.; Yang, J.; Bai, J.; Yin, M.; Cao, D.; Zhang, Q.; Zheng, L. Can circulating cell free DNA be a promising marker in ovarian cancer?—A genome-scale profiling study in a single institution. J. Ovarian Res. 2023, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.C.; Ramos, C.; Lopes-Coelho, F.; Sequeira, C.O.; Silva, F.; Gouveia-Fernandes, S.; Rodrigues, A.; Guimarães, A.; Silveira, M.; Abreu, S.; et al. Cysteine allows ovarian cancer cells to adapt to hypoxia and to escape from carboplatin cytotoxicity. Sci. Rep. 2018, 8, 9513. [Google Scholar] [CrossRef]
- Nunes, S.C.; Ramos, C.; Santos, I.; Mendes, C.; Silva, F.; Vicente, J.B.; Pereira, S.A.; Félix, A.; Gonçalves, L.G.; Serpa, J. Cysteine Boosts Fitness Under Hypoxia-Mimicked Conditions in Ovarian Cancer by Metabolic Reprogramming. Front. Cell Dev. Biol. 2021, 9, 722412. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.W.; Charkhchi, P.; Akbari, M.R. Potential clinical utility of liquid biopsies in ovarian cancer. Mol. Cancer 2022, 21, 114. [Google Scholar] [CrossRef] [PubMed]
- Trevisi, E.; Sessa, C.; Colombo, I. Clinical relevance of circulating tumor DNA in ovarian cancer: Current issues and future opportunities. Explor. Target. Antitumor. Ther. 2024, 5, 627–640. [Google Scholar] [CrossRef] [PubMed]
- de Alteriis, E.; Cartenì, F.; Parascandola, P.; Serpa, J.; Mazzoleni, S. Revisiting the Crabtree/Warburg effect in a dynamic perspective: A fitness advantage against sugar-induced cell death. Cell Cycle 2018, 17, 688–701. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal 2013, 18, 522–555. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; Park, D.-W.; Lee, H.-K.; Kim, J.-R.; Baek, S.-H. Early growth response-1 is involved in foam cell formation and is upregulated by the TLR9–MyD88–ERK1/2 pathway. Biochem. Biophys. Res. Commun. 2009, 390, 196–200. [Google Scholar] [CrossRef]
- Eruslanov, E.; Kusmartsev, S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol. Biol. 2010, 594, 57–72. [Google Scholar] [CrossRef]
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target. Ther. 2020, 5, 108. [Google Scholar] [CrossRef] [PubMed]
- Drummen, G.P.; van Liebergen, L.C.; den Kamp, J.A.O.; Post, J.A. C11-BODIPY581/591, an oxidation-sensitive fluorescent lipid peroxidation probe:(micro) spectroscopic characterization and validation of methodology. Free. Radic. Biol. Med. 2002, 33, 473–490. [Google Scholar] [CrossRef] [PubMed]
- Mitra, M.; Ho, L.D.; Coller, H.A. An in vitro Model of Cellular Quiescence in Primary Human Dermal Fibroblasts; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Truskowski, K.; Amend, S.R.; Pienta, K.J. Dormant cancer cells: Programmed quiescence, senescence, or both? Cancer Metastasis. Rev. 2023, 42, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Mungai, R.W.; Hartman, R.J.; Jolin, G.E.; Piskorowski, K.W.; Billiar, K.L. Towards a more objective and high-throughput spheroid invasion assay quantification method. Sci. Rep. 2024, 14, 31007. [Google Scholar] [CrossRef]
- Lindell, E.; Zhong, L.; Zhang, X. Quiescent Cancer Cells—A Potential Therapeutic Target to Overcome Tumor Resistance and Relapse. Int. J. Mol. Sci. 2023, 24, 3762. [Google Scholar] [CrossRef] [PubMed]
- Cvetković, D.; Goertzen, C.G.-F.; Bhattacharya, M. Quantification of breast cancer cell invasiveness using a three-dimensional (3D) model. J. Vis. Exp. JoVE 2014, 51341. [Google Scholar]
- Tevis, K.M.; Colson, Y.L.; Grinstaff, M.W. Embedded spheroids as models of the cancer microenvironment. Adv. Biosyst. 2017, 1, 1700083. [Google Scholar] [CrossRef]
- Nunes, S.C.; Lopes-Coelho, F.; Gouveia-Fernandes, S.; Ramos, C.; Pereira, S.A.; Serpa, J. Cysteine boosters the evolutionary adaptation to CoCl(2) mimicked hypoxia conditions, favouring carboplatin resistance in ovarian cancer. BMC Evol. Biol. 2018, 18, 97. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef]
- Slater, T.F. Free-radical mechanisms in tissue injury. Biochem. J. 1984, 222, 209–218. [Google Scholar] [CrossRef]
- Sugihara, K.; Nakano, S.; Koda, M.; Tanaka, K.; Fukuishi, N.; Gemba, M. Stimulatory effect of cisplatin on production of lipid peroxidation in renal tissues. Jpn. J. Pharmacol. 1987, 43, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Richez, C.; Uccellini, M.B.; Richards, R.J.; Bonegio, R.G.; Akira, S.; Monestier, M.; Corley, R.B.; Viglianti, G.A.; Marshak-Rothstein, A. Requirement for DNA CpG content in TLR9-dependent dendritic cell activation induced by DNA-containing immune complexes. J. Immunol. 2009, 183, 3109–3117. [Google Scholar] [CrossRef]
- Chockalingam, A.; Brooks, J.C.; Cameron, J.L.; Blum, L.K.; Leifer, C.A. TLR9 traffics through the Golgi complex to localize to endolysosomes and respond to CpG DNA. Immunol. Cell Biol. 2009, 87, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, G. Cancer is a propagandist. Stud. Hist. Philos. Biol. Biomed. Sci. 2017, 63, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Rich, T.A.; Reckamp, K.L.; Chae, Y.K.; Doebele, R.C.; Iams, W.T.; Oh, M.; Raymond, V.M.; Lanman, R.B.; Riess, J.W.; Stinchcombe, T.E.; et al. Analysis of Cell-Free DNA from 32,989 Advanced Cancers Reveals Novel Co-occurring Activating RET Alterations and Oncogenic Signaling Pathway Aberrations. Clin. Cancer Res. 2019, 25, 5832–5842. [Google Scholar] [CrossRef] [PubMed]
- Konkova, M.S.; Kaliyanov, A.A.; Sergeeva, V.A.; Abramova, M.S.; Kostyuk, S.V. Oxidized Cell-Free DNA Is a Factor of Stress Signaling in Radiation-Induced Bystander Effects in Different Types of Human Cells. Int. J. Genom. 2019, 2019, 9467029. [Google Scholar] [CrossRef]
- Vineis, P.; Berwick, M. The population dynamics of cancer: A Darwinian perspective. Int. J. Epidemiol. 2006, 35, 1151–1159. [Google Scholar] [CrossRef]
- Tufail, M.; Hu, J.-J.; Liang, J.; He, C.-Y.; Wan, W.-D.; Huang, Y.-Q.; Jiang, C.-H.; Wu, H.; Li, N. Hallmarks of cancer resistance. iScience 2024, 27, 109979. [Google Scholar] [CrossRef]
- Dodes Traian, M.M.; Cattoni, D.I.; Levi, V.; González Flecha, F.L. A two-stage model for lipid modulation of the activity of integral membrane proteins. PLoS ONE 2012, 7, e39255. [Google Scholar]
- Pignataro, M.F.; Dodes-Traian, M.M.; González-Flecha, F.L.; Sica, M.; Mangialavori, I.C.; Rossi, J.P.F. Modulation of plasma membrane Ca2+-ATPase by neutral phospholipids: Effect of the micelle-vesicle transition and the bilayer thickness. J. Biol. Chem. 2015, 290, 6179–6190. [Google Scholar] [PubMed]
- Parkinson, E.K.; Adamski, J.; Zahn, G.; Gaumann, A.; Flores-Borja, F.; Ziegler, C.; Mycielska, M.E. Extracellular citrate and metabolic adaptations of cancer cells. Cancer Metastasis. Rev. 2021, 40, 1073–1091. [Google Scholar] [CrossRef] [PubMed]
- Drexler, K.; Schmidt, K.M.; Jordan, K.; Federlin, M.; Milenkovic, V.M.; Liebisch, G.; Artati, A.; Schmidl, C.; Madej, G.; Tokarz, J. Cancer-associated cells release citrate to support tumour metastatic progression. Life Sci. Alliance 2021, 4. [Google Scholar]
- Lukey, M.J.; Katt, W.P.; Cerione, R.A. Targeting amino acid metabolism for cancer therapy. Drug Discov. Today 2017, 22, 796–804. [Google Scholar]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting metabolism for cancer therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar]
- Keenan, M.M.; Chi, J.-T. Alternative fuels for cancer cells. Cancer J. 2015, 21, 49–55. [Google Scholar] [PubMed]
- Green, C.R.; Wallace, M.; Divakaruni, A.S.; Phillips, S.A.; Murphy, A.N.; Ciaraldi, T.P.; Metallo, C.M. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol. 2016, 12, 15–21. [Google Scholar]
- Moffatt, B.A.; Ashihara, H. Purine and pyrimidine nucleotide synthesis and metabolism. Arab. Book/Am. Soc. Plant Biol. 2002, 1, e0018. [Google Scholar]
- Peng, H.; Wang, Y.; Luo, W. Multifaceted role of branched-chain amino acid metabolism in cancer. Oncogene 2020, 39, 6747–6756. [Google Scholar] [CrossRef]
- Chen, J.; Cui, L.; Lu, S.; Xu, S. Amino acid metabolism in tumor biology and therapy. Cell Death Dis. 2024, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Sari, I.N.; Setiawan, T.; Kim, K.S.; Wijaya, Y.T.; Cho, K.W.; Kwon, H.Y. Metabolism and function of polyamines in cancer progression. Cancer Lett. 2021, 519, 91–104. [Google Scholar]
- Ha, H.C.; Yager, J.D.; Woster, P.A.; Casero, R.A., Jr. Structural specificity of polyamines and polyamine analogues in the protection of DNA from strand breaks induced by reactive oxygen species. Biochem. Biophys. Res. Commun. 1998, 244, 298–303. [Google Scholar] [PubMed]
- Babu, B.R.; Frey, C.; Griffith, O.W. L-arginine binding to nitric-oxide synthase. The role of H-bonds to the nonreactive guanidinium nitrogens. J. Biol. Chem. 1999, 274, 25218–25226. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Tang, Q.; Zhang, K.; Huang, Q.; Ding, Y.; Jin, B.; Liu, S.; Hwa, K.; Chou, C.J.; Zhang, Y.; et al. Altered expression of the L-arginine/nitric oxide pathway in ovarian cancer: Metabolic biomarkers and biological implications. BMC Cancer 2023, 23, 844. [Google Scholar] [CrossRef]
- Daverio, Z.; Balcerczyk, A.; Rautureau, G.J.P.; Panthu, B. How Warburg-Associated Lactic Acidosis Rewires Cancer Cell Energy Metabolism to Resist Glucose Deprivation. Cancers 2023, 15, 1417. [Google Scholar] [CrossRef]
- Maeda, Y.; Kikuchi, R.; Kawagoe, J.; Tsuji, T.; Koyama, N.; Yamaguchi, K.; Nakamura, H.; Aoshiba, K. Anti-cancer strategy targeting the energy metabolism of tumor cells surviving a low-nutrient acidic microenvironment. Mol. Metab. 2020, 42, 101093. [Google Scholar] [CrossRef]
- Du, Y.; Gupta, P.; Qin, S.; Sieber, M. The role of metabolism in cellular quiescence. J. Cell Sci. 2023, 136, jcs260787. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, F.; Brand, M.D. A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 1995, 312, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.-Y.; Merl, D.; Peterson, C.W.; Wu, J.; Liu, P.Y.; Yin, H.; Muoio, D.M.; Ayer, D.E.; West, M.; Chi, J.-T. Lactic acidosis triggers starvation response with paradoxical induction of TXNIP through MondoA. PLoS Genet. 2010, 6, e1001093. [Google Scholar]
- Sivanand, S.; Vander Heiden, M.G. Emerging roles for branched-chain amino acid metabolism in cancer. Cancer Cell 2020, 37, 147–156. [Google Scholar]
- Dang, C.; Gilewski, T.A.; Surbone, A.; Norton, L. Growth curve analysis. In Holland-Frei Cancer Medicine, 6th ed.; BC Decker: Hamilton, ON, Canada, 2003. [Google Scholar]
- Toloudi, M.; Ioannou, E.; Chatziioannou, M.; Apostolou, P.; Kiritsis, C.; Manta, S.; Komiotis, D.; Papasotiriou, I. Comparison of the growth curves of cancer cells and cancer stem cells. Curr. Stem. Cell Res. Ther. 2014, 9, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef]
- Nikolaou, S.; Machesky, L.M. The stressful tumour environment drives plasticity of cell migration programmes, contributing to metastasis. J. Pathol. 2020, 250, 612–623. [Google Scholar] [CrossRef] [PubMed]
- García-Jiménez, C.; Goding, C.R. Starvation and pseudo-starvation as drivers of cancer metastasis through translation reprogramming. Cell Metab. 2019, 29, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Leong, H.C.; Datta, A.; Gopal, V.; Kumar, A.P.; Yap, C.T. PI3K/AKT signaling tips the balance of cytoskeletal forces for cancer progression. Cancers 2022, 14, 1652. [Google Scholar] [CrossRef] [PubMed]
- Pourrajab, F.; Yazdi, M.B.; Zarch, M.B.; Zarch, M.B.; Hekmatimoghaddam, S. Cross talk of the first-line defense TLRs with PI3K/Akt pathway, in preconditioning therapeutic approach. Mol. Cell. Ther. 2015, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Lima, B.H.F.; Marques, P.E.; Gomides, L.F.; Mattos, M.S.; Kraemer, L.; Queiroz-Junior, C.M.; Lennon, M.; Hirsch, E.; Russo, R.C.; Menezes, G.B. Converging TLR9 and PI3Kgamma signaling induces sterile inflammation and organ damage. Sci. Rep. 2019, 9, 19085. [Google Scholar] [CrossRef]
- Endo, H.; Owada, S.; Inagaki, Y.; Shida, Y.; Tatemichi, M. Glucose starvation induces LKB1-AMPK-mediated MMP-9 expression in cancer cells. Sci. Rep. 2018, 8, 10122. [Google Scholar] [CrossRef]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; Von Knethen, A. Nrf2, the master regulator of anti-oxidative responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef]
- Ghanem, N.; El-Baba, C.; Araji, K.; El-Khoury, R.; Usta, J.; Darwiche, N. The pentose phosphate pathway in cancer: Regulation and therapeutic opportunities. Chemotherapy 2021, 66, 179–191. [Google Scholar] [CrossRef] [PubMed]
- de Alteriis, E.; Incerti, G.; Cartenì, F.; Chiusano, M.L.; Colantuono, C.; Palomba, E.; Termolino, P.; Monticolo, F.; Esposito, A.; Bonanomi, G.; et al. Extracellular DNA secreted in yeast cultures is metabolism-specific and inhibits cell proliferation. Microb. Cell 2023, 10, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Palomba, E.; Chiusano, M.L.; Monticolo, F.; Langella, M.C.; Sanchez, M.; Tirelli, V.; de Alteriis, E.; Iannaccone, M.; Termolino, P.; Capparelli, R. Extracellular Self-DNA Effects on Yeast Cell Cycle and Transcriptome during Batch Growth. Biomolecules 2024, 14, 663. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, C.; Ling, S.; Wei, R.; Wang, J.; Xu, X. The metabolic flexibility of quiescent CSC: Implications for chemotherapy resistance. Cell Death Dis. 2021, 12, 835. [Google Scholar] [CrossRef] [PubMed]
- Steinbichler, T.B.; Savic, D.; Dudás, J.; Kvitsaridze, I.; Skvortsov, S.; Riechelmann, H.; Skvortsova, I.I. Cancer stem cells and their unique role in metastatic spread. Semin. Cancer Biol. 2020, 60, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Michelatti, D.; Beyes, S.; Bernardis, C.; Negri, M.L.; Morelli, L.; Bediaga, N.G.; Poli, V.; Fagnocchi, L.; Lago, S.; D’Annunzio, S.; et al. Oncogenic enhancers prime quiescent metastatic cells to escape NK immune surveillance by eliciting transcriptional memory. Nat. Commun. 2024, 15, 2198. [Google Scholar] [CrossRef]
- Martin, T.A.; Ye, L.; Sanders, A.J.; Lane, J.; Jiang, W.G. Cancer invasion and metastasis: Molecular and cellular perspective. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Celià-Terrassa, T.; Kang, Y. Distinctive properties of metastasis-initiating cells. Genes. Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef]
- Brasseur, K.; Gévry, N.; Asselin, E. Chemoresistance and targeted therapies in ovarian and endometrial cancers. Oncotarget 2017, 8, 4008. [Google Scholar] [CrossRef] [PubMed]
- Schnelldorfer, T.; Gansauge, S.; Gansauge, F.; Schlosser, S.; Beger, H.G.; Nussler, A.K. Glutathione depletion causes cell growth inhibition and enhanced apoptosis in pancreatic cancer cells. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2000, 89, 1440–1447. [Google Scholar] [CrossRef]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Kostyuk, S.V.; Konkova, M.S.; Ershova, E.S.; Alekseeva, A.J.; Smirnova, T.D.; Stukalov, S.V.; Kozhina, E.A.; Shilova, N.V.; Zolotukhina, T.V.; Markova, Z.G.; et al. An exposure to the oxidized DNA enhances both instability of genome and survival in cancer cells. PLoS ONE 2013, 8, e77469. [Google Scholar] [CrossRef]
- Loseva, P.; Kostyuk, S.; Malinovskaya, E.; Clement, N.; Dechesne, C.; Dani, C.; Smirnova, T.; Glebova, K.; Baidakova, G.; Baranova, A. Extracellular DNA oxidation stimulates activation of NRF2 and reduces the production of ROS in human mesenchymal stem cells. Expert. Opin. Biol. Ther. 2012, 12 (Suppl. S1), S85–S97. [Google Scholar] [PubMed]
- Filev, A.D.; Shmarina, G.V.; Ershova, E.S.; Veiko, N.N.; Martynov, A.V.; Borzikova, M.A.; Poletkina, A.A.; Dolgikh, O.A.; Veiko, V.P.; Bekker, A.A.; et al. Oxidized Cell-Free DNA Role in the Antioxidant Defense Mechanisms under Stress. Oxid. Med. Cell Longev. 2019, 2019, 1245749. [Google Scholar] [CrossRef] [PubMed]
- Combs, J.A.; DeNicola, G.M. The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers 2019, 11, 678. [Google Scholar] [CrossRef] [PubMed]
- Okuno, S.; Sato, H.; Kuriyama-Matsumura, K.; Tamba, M.; Wang, H.; Sohda, S.; Hamada, H.; Yoshikawa, H.; Kondo, T.; Bannai, S. Role of cystine transport in intracellular glutathione level and cisplatin resistance in human ovarian cancer cell lines. Br. J. Cancer 2003, 88, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Combes, A.; Camosseto, V.; N’guessan, P.; Argüello, R.J.; Mussard, J.; Caux, C.; Bendriss-Vermare, N.; Pierre, P.; Gatti, E. BAD-LAMP controls TLR9 trafficking and signalling in human plasmacytoid dendritic cells. Nat. Commun. 2017, 8, 913. [Google Scholar]
- Park, B.; Brinkmann, M.M.; Spooner, E.; Lee, C.C.; Kim, Y.M.; Ploegh, H.L. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat. Immunol. 2008, 9, 1407–1414. [Google Scholar] [CrossRef]
- Sinha, S.S.; Cameron, J.; Brooks, J.C.; Leifer, C.A. Complex negative regulation of TLR9 by multiple proteolytic cleavage events. J. Immunol. 2016, 197, 1343–1352. [Google Scholar]
- Goral, J.; Kovacs, E.J. In vivo ethanol exposure down-regulates TLR2-, TLR4-, and TLR9-mediated macrophage inflammatory response by limiting p38 and ERK1/2 activation. J. Immunol. 2005, 174, 456–463. [Google Scholar]
- Lim, E.-J.; Lee, S.-H.; Lee, J.-G.; Kim, J.-R.; Yun, S.-S.; Baek, S.-H.; Lee, C. Toll-like receptor 9 dependent activation of MAPK and NF-kB is required for the CpG ODN-induced matrix metalloproteinase-9 expression. Exp. Mol. Med. 2007, 39, 239–245. [Google Scholar] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lemos, I.; Freitas-Dias, C.; Hipólito, A.; Ramalho, J.; Carteni, F.; Gonçalves, L.G.; Mazzoleni, S.; Serpa, J. Cell-Free DNA (cfDNA) Regulates Metabolic Remodeling in the ES-2 Ovarian Carcinoma Cell Line, Influencing Cell Proliferation, Quiescence, and Chemoresistance in a Cell-of-Origin-Specific Manner. Metabolites 2025, 15, 244. https://doi.org/10.3390/metabo15040244
Lemos I, Freitas-Dias C, Hipólito A, Ramalho J, Carteni F, Gonçalves LG, Mazzoleni S, Serpa J. Cell-Free DNA (cfDNA) Regulates Metabolic Remodeling in the ES-2 Ovarian Carcinoma Cell Line, Influencing Cell Proliferation, Quiescence, and Chemoresistance in a Cell-of-Origin-Specific Manner. Metabolites. 2025; 15(4):244. https://doi.org/10.3390/metabo15040244
Chicago/Turabian StyleLemos, Isabel, Catarina Freitas-Dias, Ana Hipólito, José Ramalho, Fabrizio Carteni, Luís G. Gonçalves, Stefano Mazzoleni, and Jacinta Serpa. 2025. "Cell-Free DNA (cfDNA) Regulates Metabolic Remodeling in the ES-2 Ovarian Carcinoma Cell Line, Influencing Cell Proliferation, Quiescence, and Chemoresistance in a Cell-of-Origin-Specific Manner" Metabolites 15, no. 4: 244. https://doi.org/10.3390/metabo15040244
APA StyleLemos, I., Freitas-Dias, C., Hipólito, A., Ramalho, J., Carteni, F., Gonçalves, L. G., Mazzoleni, S., & Serpa, J. (2025). Cell-Free DNA (cfDNA) Regulates Metabolic Remodeling in the ES-2 Ovarian Carcinoma Cell Line, Influencing Cell Proliferation, Quiescence, and Chemoresistance in a Cell-of-Origin-Specific Manner. Metabolites, 15(4), 244. https://doi.org/10.3390/metabo15040244