
Influence of Amino Acids on Quorum Sensing-Related Pathways in Pseudomonas aeruginosa PAO1: Insights from the GEM iJD1249

 ,
,  ,
,  ,
,  ,
, 
Abstract

1. Introduction
2. Materials and Methods
2.1. Genome-Scale Metabolic Reconstruction of P. aeruginosa PAO1
2.2. Refinement and Curation of the GEM P. aeruginosa PAO1
2.3. Flux Balance Analysis for Metabolic Flux Distribution in GEM
2.4. Defining In Silico Growth Media and GEM Validation
2.5. Evaluation of the Metabolic Influence of Amino Acids on QS-Related Pathways
3. Results
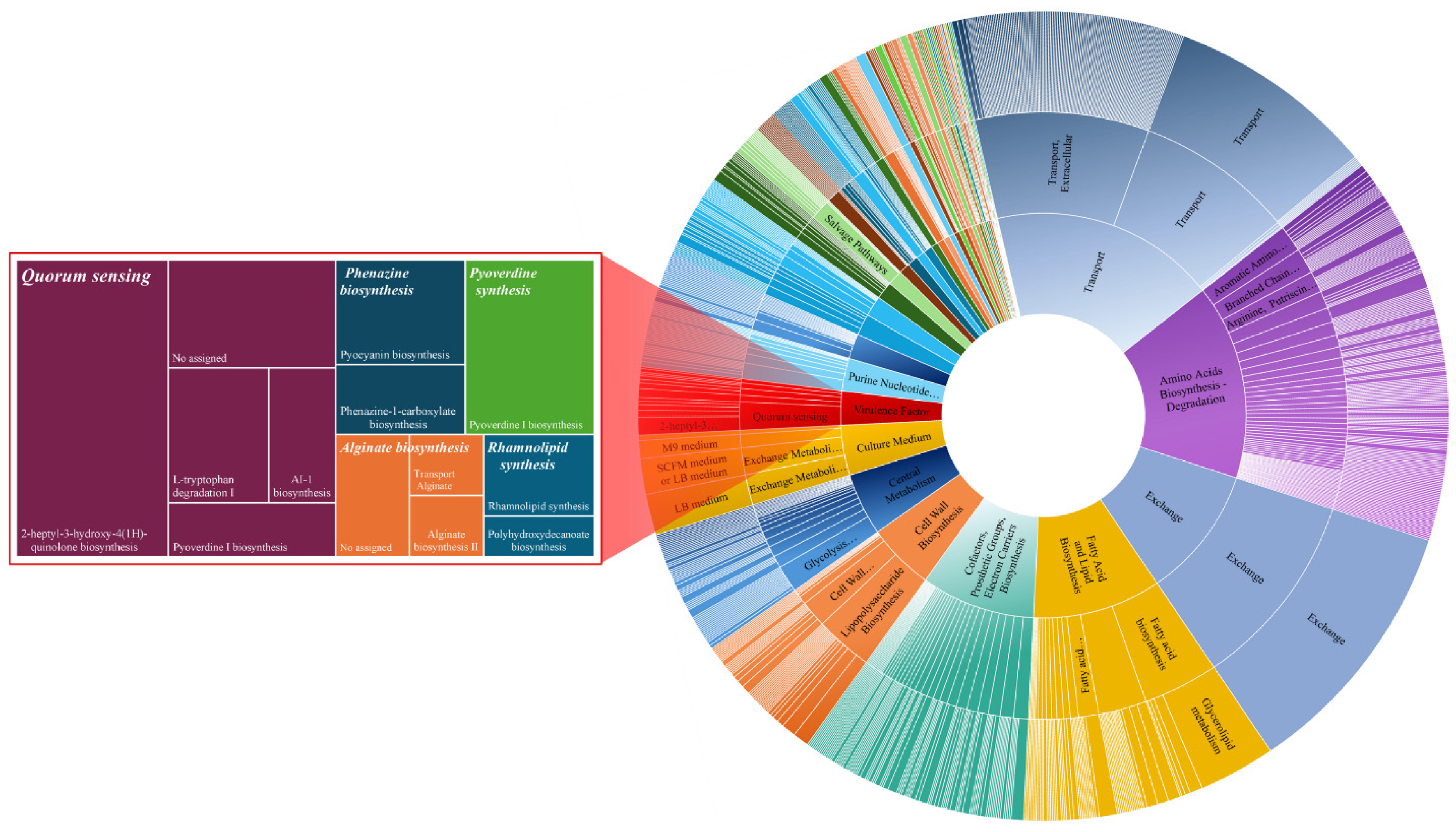
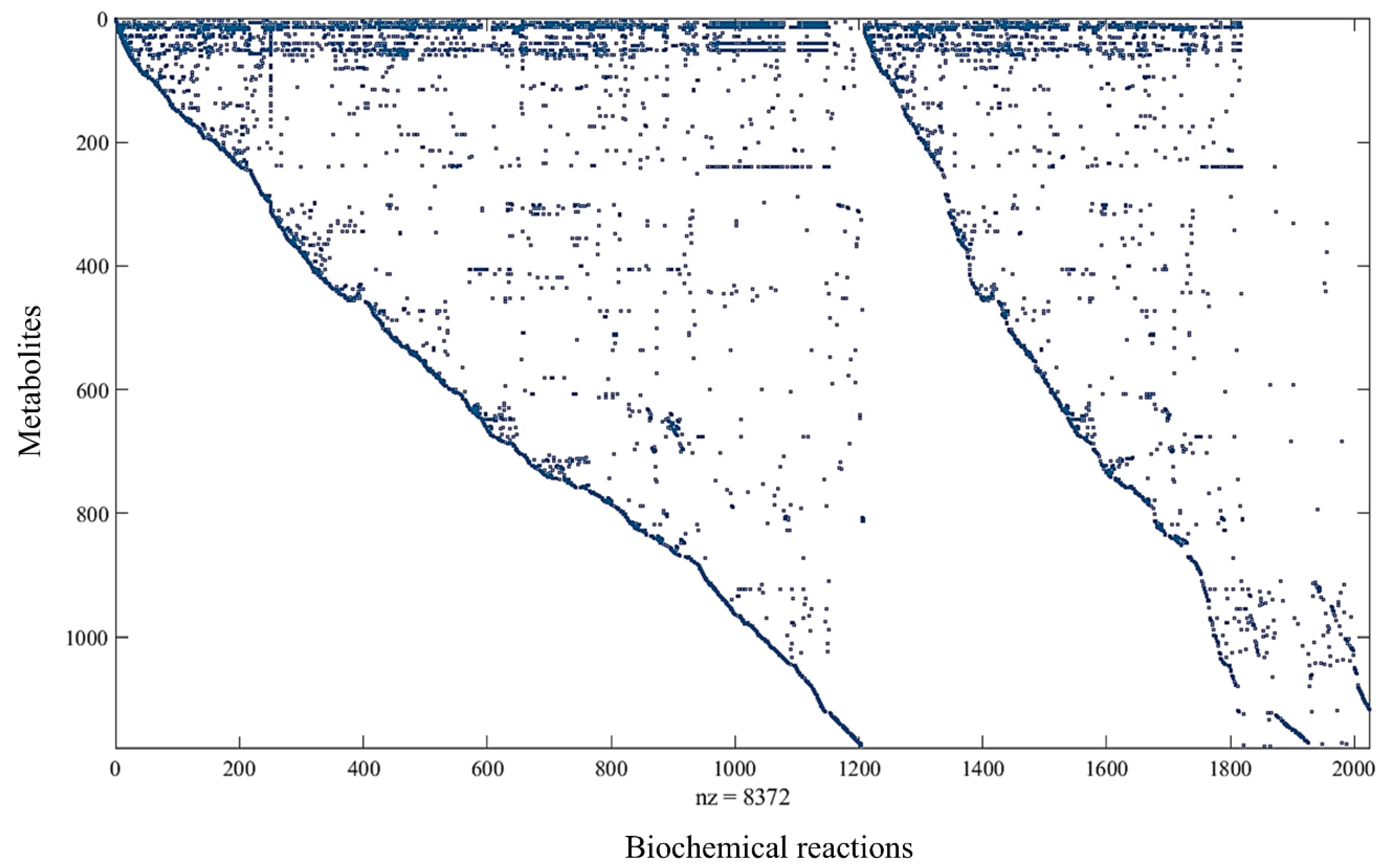
3.1. Reconstructed GEM of P. aeruginosa PAO1: iJD1246
3.2. In Silico Growth Media and GEM Validation
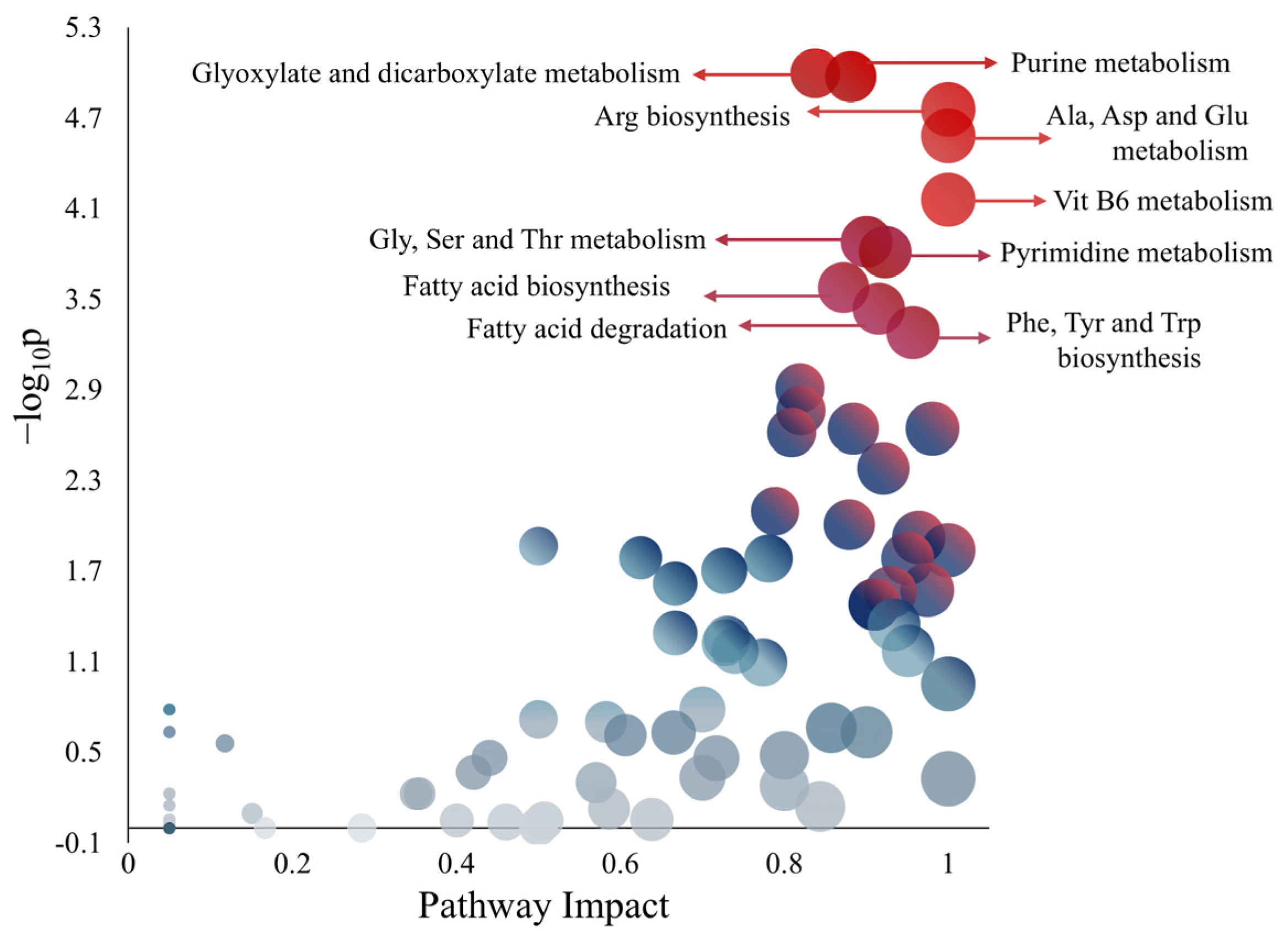
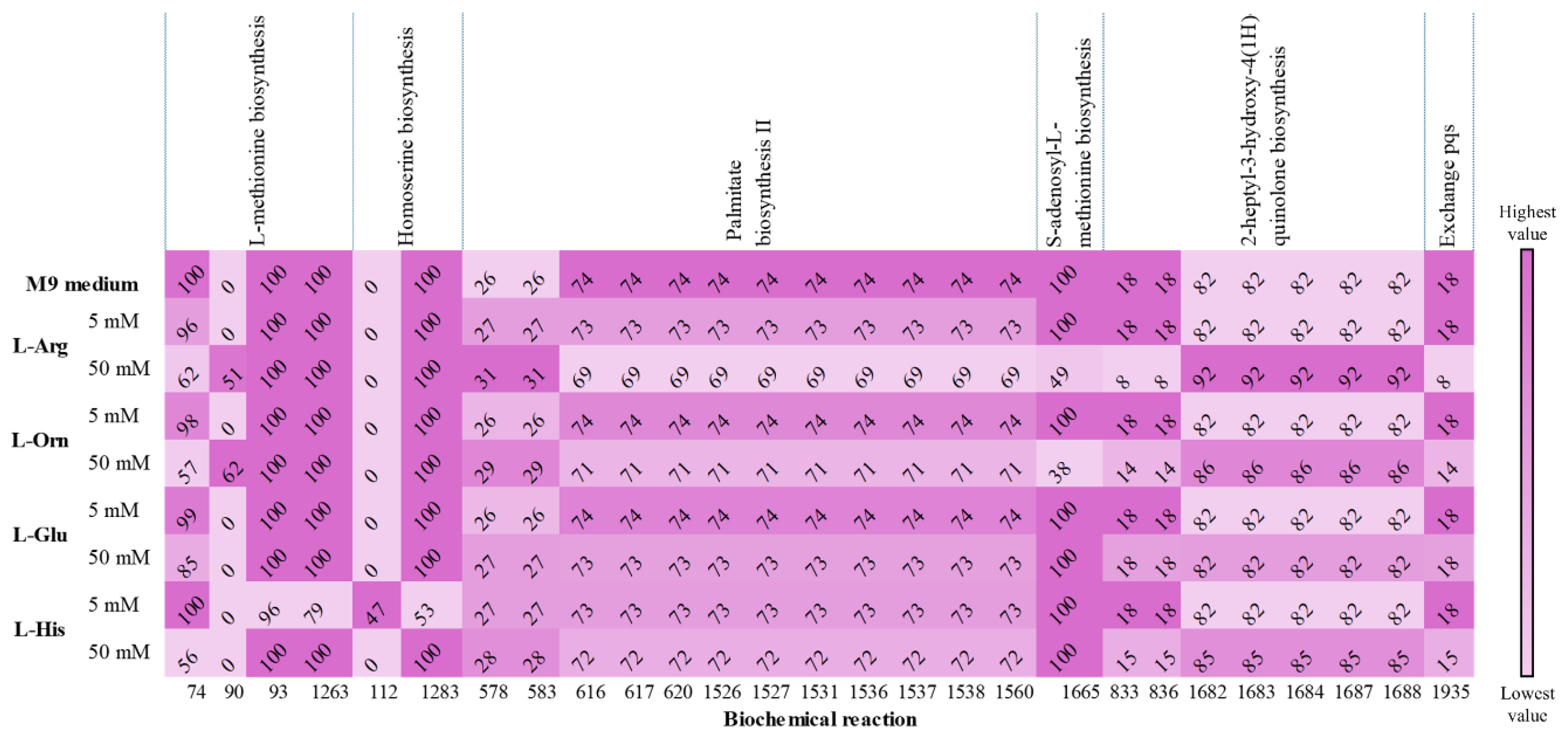
3.3. Metabolic Influence of Amino Acids on QS-Related Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAs | Amino acids |
| AHLs | Acyl-homoserine lactones |
| AIs | Autoinducers |
| butACP | Butyryl-ACP |
| 3O-C12-HSL | N-(3-oxo-dodecanoyl)-L homoserine lactone |
| C4-HSL | N-butanoyl-L-homoserine lactone |
| CF | Cystic fibrosis |
| chor | chorismate |
| FBA | Flux balance analysis |
| GEMs | Genome-scale metabolic models |
| GPR | Gene-protein-reaction |
| HHQ | 2-heptyl-3-hydroxy-4(1H)-quinolone biosynthesis |
| IQS | 2-(2-hydroxyphenyl)-thiazole-4-carbaldehyde |
| LB | Luria–Bertani |
| L-hom | L-homoserine |
| M9 | Minimal medium |
| malACP | Malonyl-ACP |
| ocCoA | octanoyl-CoA |
| ORA | Over-representation analysis |
| oxddACP | 3-oxododecanoyl-ACP |
| PPP | pentose phosphate pathway |
| PQS | 2-heptyl-3-hydroxy-4(1H)-quinolone |
| pyr | Pyruvate |
| Quorum quenching | |
| QS | Quorum sensing |
| SAM | S-adenosyl-L-methionine |
| SCFM | Synthetic cystic fibrosis medium |
| sucglu | N-Succinyl-L-glutamate |
References
- World Health Organization. Bacterial Priority Pathogens List, 2024: Bacterial Pathogens of Public Health Importance to Guide Research, Development and Strategies to Prevent and Control Antimicrobial Resistance; WHO: Geneva, Switzerland, 2024. [Google Scholar]
- Thi, M.T.T.; Wibowo, D.; Rehm, B.H.A. Pseudomonas aeruginosa Biofilms. Int. J. Mol. Sci. 2020, 21, 8671. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Huang, X.; Wang, Q.; Yao, D.; Lu, W. Virulence Factors of Pseudomonas aeruginosa and Antivirulence Strategies to Combat Its Drug Resistance. Front. Cell Infect. Microbiol. 2022, 12, 926758. [Google Scholar] [CrossRef]
- Miranda, S.W.; Asfahl, K.L.; Dandekar, A.A.; Greenberg, E.P. Pseudomonas aeruginosa Quorum Sensing. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2022; Volume 1386, pp. 95–115. [Google Scholar]
- Guo, L.; Ruan, Q.; Ma, D.; Wen, J. Revealing Quorum-Sensing Networks in Pseudomonas aeruginosa Infections through Internal and External Signals to Prevent New Resistance Trends. Microbiol. Res. 2024, 289. [Google Scholar] [CrossRef]
- Grace, A.; Sahu, R.; Owen, D.R.; Dennis, V.A. Pseudomonas aeruginosa Reference Strains PAO1 and PA14: A Genomic, Phenotypic, and Therapeutic Review. Front. Microbiol. 2022, 13, 1023523. [Google Scholar] [CrossRef]
- Liu, Y.; Ahator, S.D.; Wang, H.; Feng, Q.; Xu, Y.; Li, C.; Zhou, X.; Zhang, L.H. Microevolution of the MexT and LasR Reinforces the Bias of Quorum Sensing System in Laboratory Strains of Pseudomonas aeruginosa PAO1. Front. Microbiol. 2022, 13, 821895. [Google Scholar] [CrossRef]
- Dhodary, B.; Sampedro, I.; Behroozian, S.; Borza, V.; Her, S.; Hill, J.E. The Arginine Catabolism-Derived Amino Acid L-Ornithine Is a Chemoattractant for Pseudomonas aeruginosa. Microorganisms 2022, 10, 264. [Google Scholar] [CrossRef] [PubMed]
- Rossi, C.S.; Barrientos-Moreno, L.; Paone, A.; Cutruzzolà, F.; Paiardini, A.; Espinosa-Urgel, M.; Rinaldo, S. Nutrient Sensing and Biofilm Modulation: The Example of L-Arginine in Pseudomonas. Int. J. Mol. Sci. 2022, 23, 4386. [Google Scholar] [CrossRef]
- Abinaya, K.; Gopinath, S.C.B.; Kumarevel, T.; Raman, P. Anti-Quorum Sensing Activity of Selected Cationic Amino Acids against Chromobacterium violaceum and Pseudomonas aeruginosa. Process Biochem. 2023, 133, 75–84. [Google Scholar] [CrossRef]
- Zhang, H.; Mi, Z.; Wang, J.; Zhang, J. D-Histidine Combated Biofilm Formation and Enhanced the Effect of Amikacin against Pseudomonas aeruginosa in vitro. Arch. Microbiol. 2024, 206, 148. [Google Scholar] [CrossRef]
- Chang, R.Y.K.; Li, M.; Chow, M.Y.T.; Ke, W.R.; Tai, W.; Chan, H.K. A Dual Action of D-Amino Acids on Anti-Biofilm Activity and Moisture-Protection of Inhalable Ciprofloxacin Powders. Eur. J. Pharm. Biopharm. 2022, 173, 132–140. [Google Scholar] [CrossRef]
- Idrees, M.; Mohammad, A.R.; Karodia, N.; Rahman, A. Multimodal Role of Amino Acids in Microbial Control and Drug Development. Antibiotics 2020, 9, 330. [Google Scholar] [CrossRef] [PubMed]
- Perinbam, K.; Chacko, J.V.; Kannan, A.; Digman, M.A.; Siryaporn, A. A Shift in Central Metabolism Accompanies Virulence Activation in Pseudomonas aeruginosa. mBio 2020, 11, e02730-18. [Google Scholar] [CrossRef]
- Han, L.; Ren, J.; Xue, Y.; Gao, J.; Fu, Q.; Shao, P.; Zhu, H.; Zhang, M.; Ding, F. Fatty Acid Synthesis Promoted by PA1895-1897 Operon Delays Quorum Sensing Activation in Pseudomonas aeruginosa. AMB Express 2024, 14, 110. [Google Scholar] [CrossRef]
- Tarzi, C.; Zampieri, G.; Sullivan, N.; Angione, C. Emerging Methods for Genome-Scale Metabolic Modeling of Microbial Communities. Trends Endocrinol. Metab. 2024, 35, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Ye, A.; Shen, J.N.; Li, Y.; Lian, X.; Ma, B.G.; Guo, F.B. Reconstruction of the Genome-Scale Metabolic Network Model of Sinorhizobium Fredii CCBAU45436 for Free-Living and Symbiotic States. Front. Bioeng. Biotechnol. 2024, 12, 1377334. [Google Scholar] [CrossRef] [PubMed]
- Passi, A.; Tibocha-Bonilla, J.D.; Kumar, M.; Tec-Campos, D.; Zengler, K.; Zuniga, C. Genome-Scale Metabolic Modeling Enables in-Depth Understanding of Big Data. Metabolites 2022, 12, 14. [Google Scholar] [CrossRef]
- Turanli, B.; Gulfidan, G.; Aydogan, O.O.; Kula, C.; Selvaraj, G.; Arga, K.Y. Genome-Scale Metabolic Models in Translational Medicine: The Current Status and Potential of Machine Learning in Improving the Effectiveness of the Models. Mol. Omics 2024, 20, 234–247. [Google Scholar] [CrossRef]
- Domenzain, I.; Sánchez, B.; Anton, M.; Kerkhoven, E.J.; Millán-Oropeza, A.; Henry, C.; Siewers, V.; Morrissey, J.P.; Sonnenschein, N.; Nielsen, J. Reconstruction of a Catalogue of Genome-Scale Metabolic Models with Enzymatic Constraints Using GECKO 2.0. Nat. Commun. 2022, 13, 3766. [Google Scholar] [CrossRef]
- Marin de Mas, I.; Herand, H.; Carrasco, J.; Nielsen, L.K.; Johansson, P.I. A Protocol for the Automatic Construction of Highly Curated Genome-Scale Models of Human Metabolism. Bioengineering 2023, 10, 576. [Google Scholar] [CrossRef]
- Bi, X.; Liu, Y.; Li, J.; Du, G.; Lv, X.; Liu, L. Construction of Multiscale Genome-Scale Metabolic Models: Frameworks and Challenges. Biomolecules 2022, 12, 721. [Google Scholar] [CrossRef]
- Oberhardt, M.A.; Puchałka, J.; Fryer, K.E.; Martins Dos Santos, V.A.P.; Papin, J.A. Genome-Scale Metabolic Network Analysis of the Opportunistic Pathogen Pseudomonas aeruginosa PAO1. J. Bacteriol. 2008, 190, 2790–2803. [Google Scholar] [CrossRef] [PubMed]
- Bartell, J.A.; Blazier, A.S.; Yen, P.; Thøgersen, J.C.; Jelsbak, L.; Goldberg, J.B.; Papin, J.A. Reconstruction of the Metabolic Network of Pseudomonas aeruginosa to Interrogate Virulence Factor Synthesis. Nat. Commun. 2017, 8, 14631. [Google Scholar] [CrossRef]
- Clavijo-Buriticá, D.C.; Arévalo-Ferro, C.; González Barrios, A.F. A Holistic Approach from Systems Biology Reveals the Direct Influence of the Quorum-Sensing Phenomenon on Pseudomonas aeruginosa Metabolism to Pyoverdine Biosynthesis. Metabolites 2023, 13, 659. [Google Scholar] [CrossRef] [PubMed]
- Seif, Y.; Palsson, B.Ø. Path to Improving the Life Cycle and Quality of Genome-Scale Models of Metabolism. Cell Syst. 2021, 12, 842–859. [Google Scholar] [PubMed]
- Thiele, I.; Palsson, B. A Protocol for Generating a High-Quality Genome-Scale Metabolic Reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef]
- Norsigian, C.J.; Fang, X.; Seif, Y.; Monk, J.M.; Palsson, B.O. A Workflow for Generating Multi-Strain Genome-Scale Metabolic Models of Prokaryotes. Nat. Protoc. 2020, 15, 1–14. [Google Scholar] [CrossRef]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc Collection of Microbial Genomes and Metabolic Pathways. Brief. Bioinform. 2018, 20, 1085–1093. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for Taxonomy-Based Analysis of Pathways and Genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR Core Data Resource in 2021: New Developments and Updates. Nucleic Acids Res 2021, 49, D498–D508. [Google Scholar] [CrossRef]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as Designed by Its Users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef]
- McDonald, A.G.; Boyce, S.; Tipton, K.F. ExplorEnz: The Primary Source of the IUBMB Enzyme List. Nucleic Acids Res. 2009, 37, D593–D597. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; Cukura, A.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Bansal, P.; Morgat, A.; Axelsen, K.B.; Muthukrishnan, V.; Coudert, E.; Aimo, L.; Hyka-Nouspikel, N.; Gasteiger, E.; Kerhornou, A.; Neto, T.B.; et al. Rhea, the Reaction Knowledgebase in 2022. Nucleic Acids Res. 2022, 50, D693–D700. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Tran, V.D.T.; Mehl, F.; Ibberson, M.; Pagni, M. MetaNetX/MNXref: Unified Namespace for Metabolites and Biochemical Reactions in the Context of Metabolic Models. Nucleic Acids Res. 2021, 49, D570–D574. [Google Scholar] [CrossRef]
- Pang, Z.; Lu, Y.; Zhou, G.; Hui, F.; Xu, L.; Viau, C.; Spigelman, A.F.; Macdonald, P.E.; Wishart, D.S.; Li, S.; et al. MetaboAnalyst 6.0: Towards a Unified Platform for Metabolomics Data Processing, Analysis and Interpretation. Nucleic Acids Res. 2024, 52, W398–W406. [Google Scholar] [CrossRef] [PubMed]
- Wieder, C.; Frainay, C.; Poupin, N.; Rodríguez-Mier, P.; Vinson, F.; Cooke, J.; Lai, R.P.J.; Bundy, J.G.; Jourdan, F.; Ebbels, T. Pathway Analysis in Metabolomics: Recommendations for the Use of over-Representation Analysis. PLoS Comput. Biol. 2021, 17, e1009105. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef]
- Padilla-De La-Rosa, J.D.; Garćia-Raḿirez, M.A.; Gschaedler-Mathis, A.C.; Ǵomez-Guzḿan, A.I.; Soĺis-Pacheco, J.R.; Gonźalez-Reynoso, O. Estimation of Metabolic Fluxes Distribution in Saccharomyces cerevisiae during the Production of Volatile Compounds of Tequila. Math. Biosci. Eng. 2021, 18, 5094–5113. [Google Scholar] [CrossRef]
- Marinos, G.; Kaleta, C.; Waschina, S. Defining the Nutritional Input for Genome-Scale Metabolic Models: A Roadmap. PLoS ONE 2020, 15, e0236890. [Google Scholar] [CrossRef]
- Oberhardt, M.A.; Goldberg, J.B.; Hogardt, M.; Papin, J.A. Metabolic Network Analysis of Pseudomonas aeruginosa during Chronic Cystic Fibrosis Lung Infection. J. Bacteriol. 2010, 192, 5534–5548. [Google Scholar] [CrossRef]
- Reed, J.L.; Vo, T.D.; Schilling, C.H.; Palsson, B.O. An Expanded Genome-Scale Model of Escherichia coli K-12 (IJR904 GSM/GPR). Genome Biol. 2003, 4, R54. [Google Scholar]
- Liu, Y.K.; Kuo, H.C.; Lai, C.H.; Chou, C.C. Single Amino Acid Utilization for Bacterial Categorization. Sci. Rep. 2020, 10, 12686. [Google Scholar] [CrossRef]
- Dubern, J.F.; Halliday, N.; Cámara, M.; Winzer, K.; Barrett, D.A.; Hardie, K.R.; Williams, P. Growth Rate and Nutrient Limitation as Key Drivers of Extracellular Quorum Sensing Signal Molecule Accumulation in Pseudomonas aeruginosa. Microbiology 2023, 169, 001316. [Google Scholar] [CrossRef]
- Berger, A.; Dohnt, K.; Tielen, P.; Jahn, D.; Becker, J.; Wittmann, C. Robustness and Plasticity of Metabolic Pathway Flux among Uropathogenic Isolates of Pseudomonas aeruginosa. PLoS ONE 2014, 9, e88368. [Google Scholar] [CrossRef]
- Opperman, M.J.; Shachar-Hill, Y. Metabolic Flux Analyses of Pseudomonas aeruginosa Cystic Fibrosis Isolates. Metab. Eng. 2016, 38, 251–263. [Google Scholar] [CrossRef]
- Milo, R.; Jorgensen, P.; Moran, U.; Weber, G.; Springer, M. BioNumbers The Database of Key Numbers in Molecular and Cell Biology. Nucleic Acids Res. 2010, 38, D750–D753. [Google Scholar] [CrossRef]
- Ruhluel, D.; O’Brien, S.; Fothergill, J.L.; Neill, D.R. Development of Liquid Culture Media Mimicking the Conditions of Sinuses and Lungs in Cystic Fibrosis and Health. F1000Res 2022, 11, 1007. [Google Scholar] [CrossRef]
- Palmer, K.L.; Aye, L.M.; Whiteley, M. Nutritional Cues Control Pseudomonas aeruginosa Multicellular Behavior in Cystic Fibrosis Sputum. J. Bacteriol. 2007, 189, 8079–8087. [Google Scholar] [CrossRef]
- Espaillat, A.; Colque, C.A.; Rago, D.; Rosa, R.L.; Molin, S.; Johansen, H.K. Adaptive Evolution of Pseudomonas aeruginosa in Human Airways Shows Phenotypic Convergence Despite Diverse Patterns of Genomic Changes. Mol. Biol. Evol. 2024, 41, msae022. [Google Scholar] [CrossRef]
- De Plano, L.M.; Caratozzolo, M.; Conoci, S.; Guglielmino, S.P.P.; Franco, D. Impact of Nutrient Starvation on Biofilm Formation in Pseudomonas aeruginosa: An Analysis of Growth, Adhesion, and Spatial Distribution. Antibiotics 2024, 13, 987. [Google Scholar] [CrossRef]
- Wijesinghe, G.; Dilhari, A.; Gayani, B.; Kottegoda, N.; Samaranayake, L.; Weerasekera, M. Influence of Laboratory Culture Media on in Vitro Growth, Adhesion, and Biofilm Formation of Pseudomonas aeruginosa and Staphylococcus aureus. Med. Princ. Pract. 2019, 28, 28–35. [Google Scholar] [CrossRef]
- Kao, W.T.K.; Frye, M.; Gagnon, P.; Vogel, J.P.; Chole, R. D-Amino Acids Do Not Inhibit Pseudomonas aeruginosa Biofilm Formation. Laryngoscope Investig. Otolaryngol. 2017, 2, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Rumbo, C.; Vallejo, J.A.; Cabral, M.P.; Martínez-Guitián, M.; Pérez, A.; Beceiro, A.; Bou, G. Assessment of Antivirulence Activity of Several D-Amino Acids against Acinetobacter baumannii and Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2016, 71, 3473–3481. [Google Scholar] [CrossRef]
- Bin, P.; Zhu, C.; Liu, S.; Li, Z.; Ren, W.; Zhu, G. Perspective: Methionine Restriction–Induced Longevity—A Possible Role for Inhibiting the Synthesis of Bacterial Quorum Sensing Molecules. Adv. Nutr. 2020, 11, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Joseph, C.; Zafeiropoulos, H.; Bernaerts, K.; Faust, K. Predicting Microbial Interactions with Approaches Based on Flux Balance Analysis: An Evaluation. BMC Bioinform. 2024, 25, 36. [Google Scholar] [CrossRef] [PubMed]
- Eisha, S.; Morris, A.J.; Martin, I.; Yau, Y.C.W.; Grasemann, H.; Waters, V. Effect of L-Arginine on Cystic Fibrosis Pseudomonas aeruginosa Biofilms. Antimicrob. Agents Chemother. 2024, 68, e0033624. [Google Scholar] [CrossRef]
- Morris, C.R.; Hamilton-Reeves, J.; Martindale, R.G.; Sarav, M.; Ochoa Gautier, J.B. Acquired Amino Acid Deficiencies: A Focus on Arginine and Glutamine. Nutr. Clin. Pract. 2017, 32, 30S–47S. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Proteins | Biochemical Reactions | Exchange Reactions | Metabolites | Compartments | |
|---|---|---|---|---|---|---|
| iJD1249 | 1249 | 1051 | 1208 | 205 | 1178 | 3 a |
| iPae1146 [24] | 1146 | 22 | 1321 | 172 | 1284 | 2 b |
| CCBM1146 [25] | 1146 | 1009 | 1123 | 120 | 880 | 2 b |
| iMO1056 [23] | 1056 | 1030 | 883 | 118 | 760 | 2 b |
| Medium | µ (h−1) | td (h) |
|---|---|---|
| M9 (4 gL−1 glucose) | 0.4016 | 1.7260 |
| SCFM | 0.4055 | 1.7094 |
| LB | 0.6943 | 0.9983 |
| µ (h−1) | ||
|---|---|---|
| Amino Acid | 5 mM | 50 mM |
| D-Met | 0.4016 | 0.4016 |
| D-Ala | 0.4236 | 0.6211 |
| D-Glu | 0.4236 | 0.6171 |
| D-Ser | 0.4236 | 0.6047 |
| L-Orn | 0.4455 | 0.7583 |
| L-His | 0.4675 | 1.0041 |
| L-Glu | 0.4236 | 0.6171 |
| L-Arg | 0.4894 | 1.0223 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgado-Nungaray, J.A.; Figueroa-Yáñez, L.J.; Reynaga-Delgado, E.; García-Ramírez, M.A.; Aguilar-Corona, K.E.; Gonzalez-Reynoso, O. Influence of Amino Acids on Quorum Sensing-Related Pathways in Pseudomonas aeruginosa PAO1: Insights from the GEM iJD1249. Metabolites 2025, 15, 236. https://doi.org/10.3390/metabo15040236
Delgado-Nungaray JA, Figueroa-Yáñez LJ, Reynaga-Delgado E, García-Ramírez MA, Aguilar-Corona KE, Gonzalez-Reynoso O. Influence of Amino Acids on Quorum Sensing-Related Pathways in Pseudomonas aeruginosa PAO1: Insights from the GEM iJD1249. Metabolites. 2025; 15(4):236. https://doi.org/10.3390/metabo15040236
Chicago/Turabian StyleDelgado-Nungaray, Javier Alejandro, Luis Joel Figueroa-Yáñez, Eire Reynaga-Delgado, Mario Alberto García-Ramírez, Karla Esperanza Aguilar-Corona, and Orfil Gonzalez-Reynoso. 2025. "Influence of Amino Acids on Quorum Sensing-Related Pathways in Pseudomonas aeruginosa PAO1: Insights from the GEM iJD1249" Metabolites 15, no. 4: 236. https://doi.org/10.3390/metabo15040236
APA StyleDelgado-Nungaray, J. A., Figueroa-Yáñez, L. J., Reynaga-Delgado, E., García-Ramírez, M. A., Aguilar-Corona, K. E., & Gonzalez-Reynoso, O. (2025). Influence of Amino Acids on Quorum Sensing-Related Pathways in Pseudomonas aeruginosa PAO1: Insights from the GEM iJD1249. Metabolites, 15(4), 236. https://doi.org/10.3390/metabo15040236