
Cell-Free DNA (cfDNA) Regulates Metabolic Remodeling, Sustaining Proliferation, Quiescence, and Migration in MDA-MB-231, a Triple-Negative Breast Carcinoma (TNBC) Cell Line

 , , ,
, , ,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture Conditions
2.2. Cell-Free DNA Isolation
2.3. Cell Proliferation
2.4. Wound Healing Assay
2.5. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.6. Cell Death Analysis by Flow Cytometry
2.7. Reactive Oxygen Species (ROS) Quantification by Flow Cytometry
2.8. Lipid Peroxide Quantification by Flow Cytometry
2.9. Immunofluorescence
2.10. Lentivirus Transduction
2.11. Co-Culture Growth Advantage Assay
2.12. Statistical Analysis
3. Results
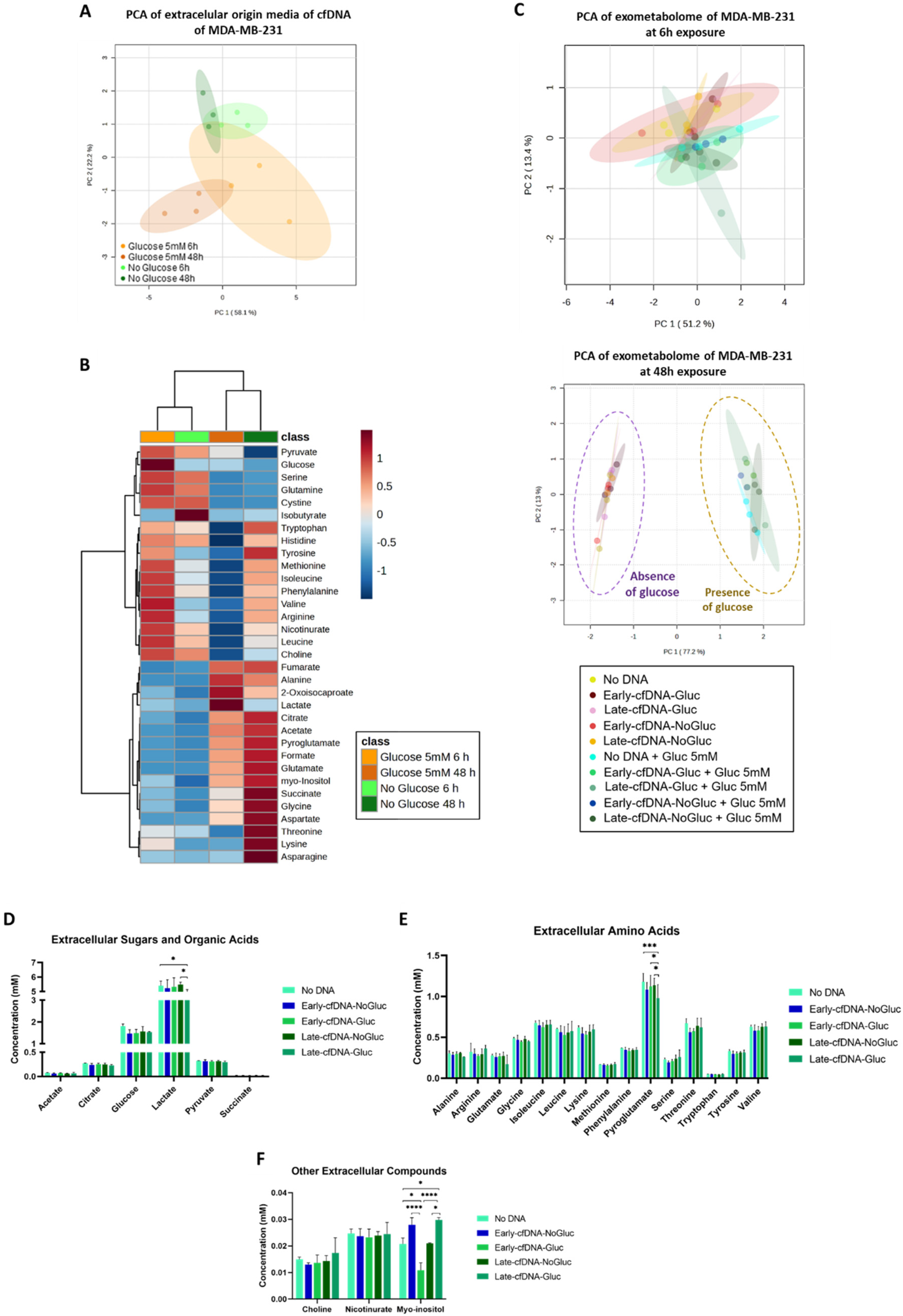
3.1. The Consumption of Glucose by MDA-MB-231 Is Significantly Affected by cfDNA Exposure
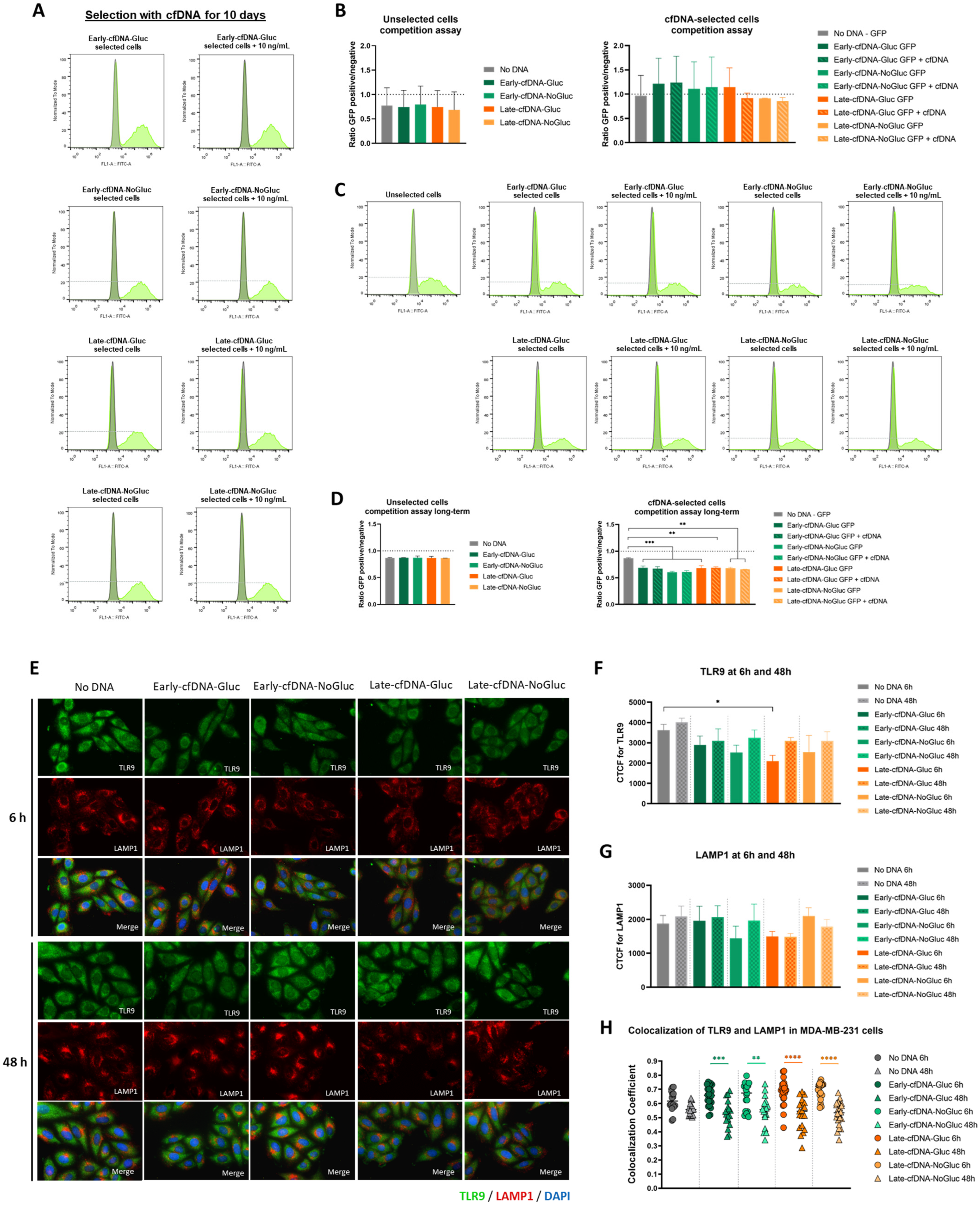
3.2. Late-cfDNA-NoGluc and Late-cfDNA-Gluc Tended to Decrease MDA-MB-231 Cell Death Levels and Increase Migration in the Presence of Glucose
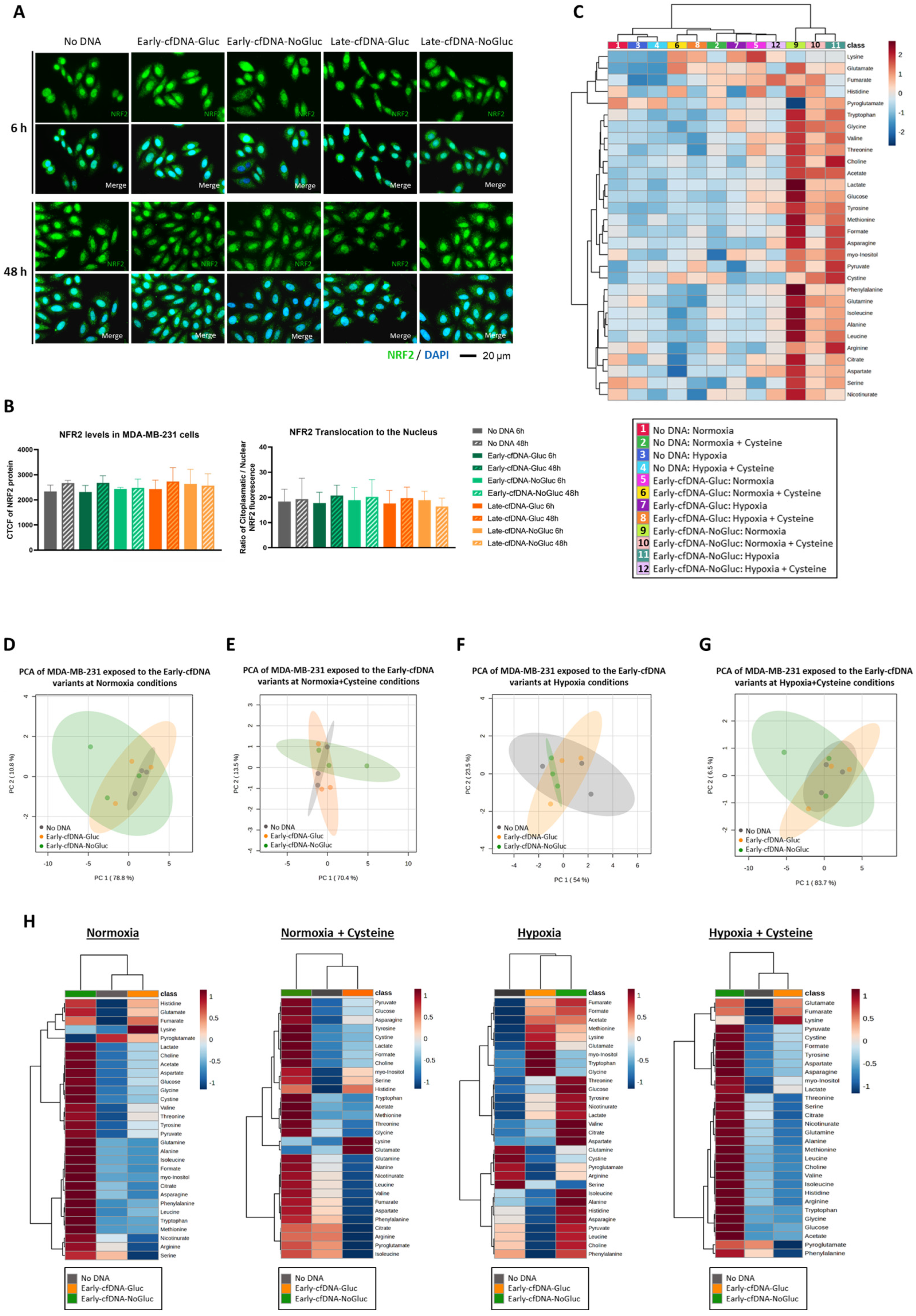
3.3. Early-cfDNA-Gluc and Late-cfDNA-Gluc Promoted Cisplatin Resistance in MDA-MB-231 Cells, Which Is Favored by Hypoxia and Cysteine Availability
3.4. Cysteine Enhances the Protection of MDA-MB-231 Cells from Oxidative Damage by Early-cfDNA-Gluc and Late-cfDNA-Gluc
3.5. The Early-cfDNA-NoGluc Variant Induced a Metabolic Remodeling to Support Chemoresistance
3.6. Long-Term Selection with Late-cfDNA-NoGluc and Late-cfDNA-Gluc Variants Induced Quiescence in MDA-MB-231 Cells
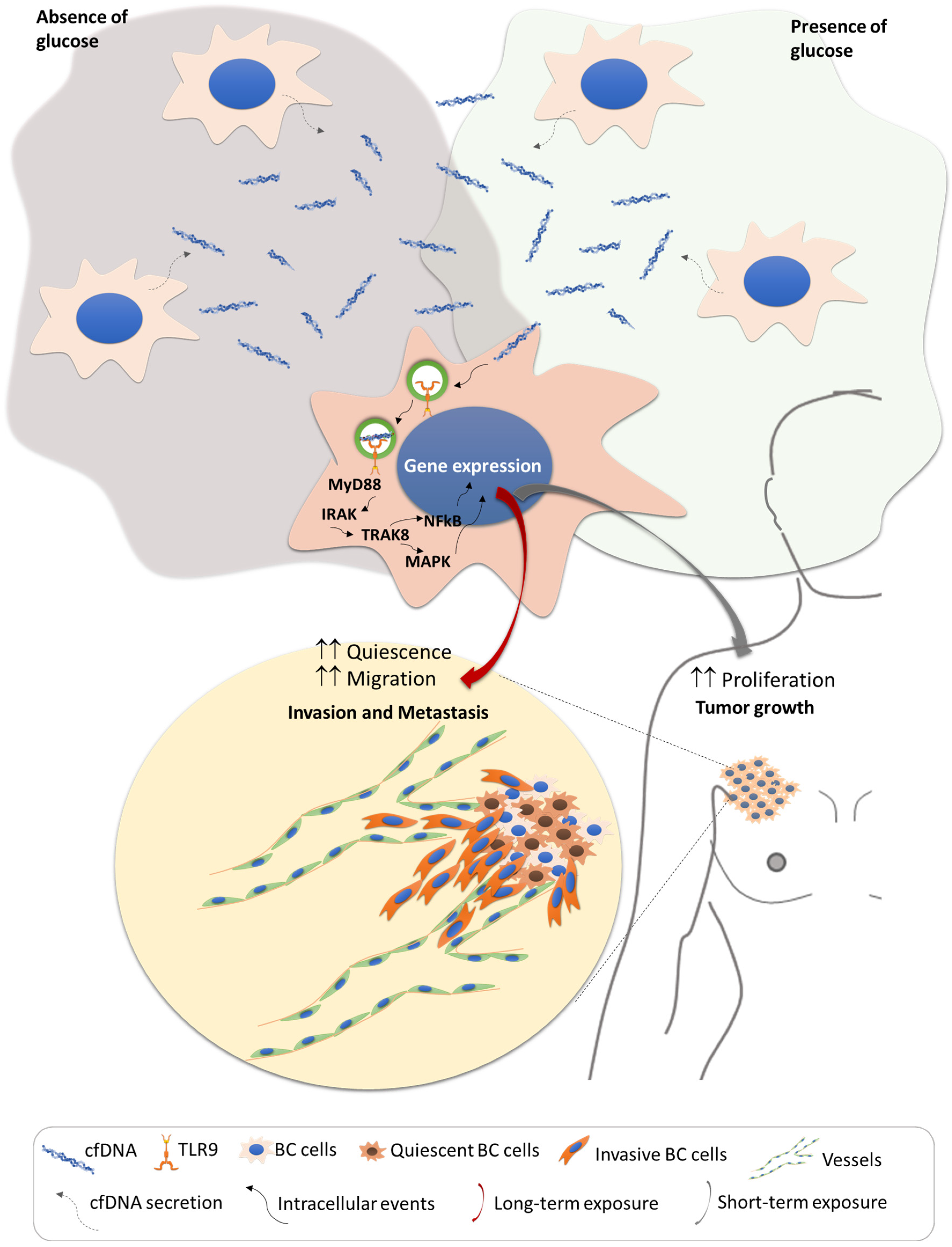
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aucamp, J.; Bronkhorst, A.J.; Badenhorst, C.P.S.; Pretorius, P.J. The diverse origins of circulating cell-free DNA in the human body: A critical re-evaluation of the literature. Biol. Rev. Camb. Philos. Soc. 2018, 93, 1649–1683. [Google Scholar] [CrossRef] [PubMed]
- Canzoniero, J.V.; Park, B.H. Use of cell free DNA in breast oncology. Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 2016, 1865, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Duvvuri, B.; Lood, C. Cell-Free DNA as a Biomarker in Autoimmune Rheumatic Diseases. Front. Immunol. 2019, 10, 502. [Google Scholar] [CrossRef]
- Trulson, I.; Stahl, J.; Margraf, S.; Scholz, M.; Hoecherl, E.; Wolf, K.; Durner, J.; Klawonn, F.; Holdenrieder, S. Cell-free DNA in plasma and serum indicates disease severity and prognosis in blunt trauma patients. Diagnostics 2023, 13, 1150. [Google Scholar] [CrossRef]
- Urosevic, N.; Merritt, A.J.; Inglis, T.J. Plasma cfDNA predictors of established bacteraemic infection. Access Microbiol. 2022, 4, 000373. [Google Scholar] [CrossRef]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar]
- Atamaniuk, J.; Vidotto, C.; Kinzlbauer, M.; Bachl, N.; Tiran, B.; Tschan, H. Cell-free plasma DNA and purine nucleotide degradation markers following weightlifting exercise. Eur. J. Appl. Physiol. 2010, 110, 695–701. [Google Scholar] [CrossRef]
- Butt, A.N.; Swaminathan, R. Overview of circulating nucleic acids in plasma/serum: Update on potential prognostic and diagnostic value in diseases excluding fetal medicine and oncology. Ann. N. Y. Acad. Sci. 2008, 1137, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Volik, S.; Alcaide, M.; Morin, R.D.; Collins, C. Cell-free DNA (cfDNA): Clinical Significance and Utility in Cancer Shaped By Emerging Technologies. Mol. Cancer Res. 2016, 14, 898–908. [Google Scholar] [CrossRef]
- Fettke, H.; Kwan, E.M.; Azad, A.A. Cell-free DNA in cancer: Current insights. Cell Oncol. 2019, 42, 13–28. [Google Scholar] [CrossRef]
- Khakoo, S.; Georgiou, A.; Gerlinger, M.; Cunningham, D.; Starling, N. Circulating tumour DNA, a promising biomarker for the management of colorectal cancer. Crit. Rev. Oncol. Hematol. 2018, 122, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, S.; Machesky, L.M. The stressful tumour environment drives plasticity of cell migration programmes, contributing to metastasis. J. Pathol. 2020, 250, 612–623. [Google Scholar]
- García-Jiménez, C.; Goding, C.R. Starvation and pseudo-starvation as drivers of cancer metastasis through translation reprogramming. Cell Metab. 2019, 29, 254–267. [Google Scholar] [PubMed]
- Deng, S.; Leong, H.C.; Datta, A.; Gopal, V.; Kumar, A.P.; Yap, C.T. PI3K/AKT signaling tips the balance of cytoskeletal forces for cancer progression. Cancers 2022, 14, 1652. [Google Scholar] [CrossRef]
- Yan, Y.-y.; Guo, Q.-r.; Wang, F.-h.; Adhikari, R.; Zhu, Z.-y.; Zhang, H.-y.; Zhou, W.-m.; Yu, H.; Li, J.-q.; Zhang, J.-y. Cell-Free DNA: Hope and Potential Application in Cancer. Front. Cell Dev. Biol. 2021, 9, 639233. [Google Scholar] [CrossRef]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112.e114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liang, Y.; Li, S.; Zeng, F.; Meng, Y.; Chen, Z.; Liu, S.; Tao, Y.; Yu, F. The interplay of circulating tumor DNA and chromatin modification, therapeutic resistance, and metastasis. Mol. Cancer 2019, 18, 36. [Google Scholar] [CrossRef]
- Orrantia-Borunda, E.; Anchondo-Nuñez, P.; Acuña-Aguilar, L.E.; Gómez-Valles, F.O.; Ramírez-Valdespino, C.A. Subtypes of Breast Cancer, in Breast Cancer; Mayrovitz, H.N., Ed.; Exon Publications: Brisbane, AU, USA, 2022. [Google Scholar]
- Shaath, H.; Elango, R.; Alajez, N.M. Molecular classification of breast cancer utilizing long non-coding RNA (lncRNA) transcriptomes identifies novel diagnostic lncRNA panel for triple-negative breast cancer. Cancers 2021, 13, 5350. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar]
- Elhelaly, R.; Effat, N.; Hegazy, M.A.E.-F.; Abdelwahab, K.; Hamdy, O.; Hashem, E.M.A.; Elzehery, R.R. Circulating cell free DNA and DNA integrity index as discriminating tools between breast cancer and benign breast disease. Asian Pac. J. Cancer Prev. APJCP 2022, 23, 545. [Google Scholar]
- Teo, Y.V.; Capri, M.; Morsiani, C.; Pizza, G.; Faria, A.M.C.; Franceschi, C.; Neretti, N. Cell-free DNA as a biomarker of aging. Aging Cell 2019, 18, e12890. [Google Scholar] [PubMed]
- Pushpanjali, P.; Keshari, J.; Prakash, P.; Kumar, M.; Mandal, M.; Kumari, R. Correlation between circulating cell-free DNA levels and breast cancer subtypes: A prospective observational study. Cureus 2023, 15, e42247. [Google Scholar]
- Ferrari, P.; Scatena, C.; Ghilli, M.; Bargagna, I.; Lorenzini, G.; Nicolini, A. Molecular mechanisms, biomarkers and emerging therapies for chemotherapy resistant TNBC. Int. J. Mol. Sci. 2022, 23, 1665. [Google Scholar] [CrossRef]
- Brown, L.C.; Salgado, R.; Luen, S.J.; Savas, P.; Loi, S. Tumor-infiltrating lymphocyctes in triple-negative breast cancer: Update for 2020. Cancer J. 2021, 27, 25–31. [Google Scholar] [PubMed]
- Won, K.A.; Spruck, C. Triple-negative breast cancer therapy: Current and future perspectives. Int. J. Oncol. 2020, 57, 1245–1261. [Google Scholar] [CrossRef]
- O′Reilly, D.; Al Sendi, M.; Kelly, C.M. Overview of recent advances in metastatic triple negative breast cancer. World J. Clin. Oncol. 2021, 12, 164. [Google Scholar] [PubMed]
- Nunes, S.C.; Ramos, C.; Lopes-Coelho, F.; Sequeira, C.O.; Silva, F.; Gouveia-Fernandes, S.; Rodrigues, A.; Guimarães, A.; Silveira, M.; Abreu, S.; et al. Cysteine allows ovarian cancer cells to adapt to hypoxia and to escape from carboplatin cytotoxicity. Sci. Rep. 2018, 8, 9513. [Google Scholar] [CrossRef]
- Nunes, S.C.; Ramos, C.; Santos, I.; Mendes, C.; Silva, F.; Vicente, J.B.; Pereira, S.A.; Félix, A.; Gonçalves, L.G.; Serpa, J. Cysteine Boosts Fitness Under Hypoxia-Mimicked Conditions in Ovarian Cancer by Metabolic Reprogramming. Front. Cell Dev. Biol. 2021, 9, 722412. [Google Scholar] [CrossRef]
- Nunes, S.C.; Lopes-Coelho, F.; Gouveia-Fernandes, S.; Ramos, C.; Pereira, S.A.; Serpa, J. Cysteine boosters the evolutionary adaptation to CoCl(2) mimicked hypoxia conditions, favouring carboplatin resistance in ovarian cancer. BMC Evol. Biol. 2018, 18, 97. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef] [PubMed]
- Slater, T.F. Free-radical mechanisms in tissue injury. Biochem. J. 1984, 222, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, K.; Nakano, S.; Koda, M.; Tanaka, K.; Fukuishi, N.; Gemba, M. Stimulatory effect of cisplatin on production of lipid peroxidation in renal tissues. Jpn. J. Pharmacol. 1987, 43, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Rustamov, N.; Roh, Y.S. The Roles of NFR2-Regulated Oxidative Stress and Mitochondrial Quality Control in Chronic Liver Diseases. Antioxidants 2023, 12, 1928. [Google Scholar] [CrossRef]
- Zang, H.; Mathew, R.O.; Cui, T. The Dark Side of Nrf2 in the Heart. Front. Physiol. 2020, 11, 722. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef]
- Muinelo-Romay, L.; Casas-Arozamena, C.; Abal, M. Liquid Biopsy in Endometrial Cancer: New Opportunities for Personalized Oncology. Int. J. Mol. Sci. 2018, 19, 2311. [Google Scholar] [CrossRef]
- Rich, T.A.; Reckamp, K.L.; Chae, Y.K.; Doebele, R.C.; Iams, W.T.; Oh, M.; Raymond, V.M.; Lanman, R.B.; Riess, J.W.; Stinchcombe, T.E.; et al. Analysis of Cell-Free DNA from 32,989 Advanced Cancers Reveals Novel Co-occurring Activating RET Alterations and Oncogenic Signaling Pathway Aberrations. Clin. Cancer Res. 2019, 25, 5832–5842. [Google Scholar] [CrossRef]
- Konkova, M.S.; Kaliyanov, A.A.; Sergeeva, V.A.; Abramova, M.S.; Kostyuk, S.V. Oxidized Cell-Free DNA Is a Factor of Stress Signaling in Radiation-Induced Bystander Effects in Different Types of Human Cells. Int. J. Genom. 2019, 2019, 9467029. [Google Scholar] [CrossRef]
- Lukey, M.J.; Katt, W.P.; Cerione, R.A. Targeting amino acid metabolism for cancer therapy. Drug Discov. Today 2017, 22, 796–804. [Google Scholar] [CrossRef]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting metabolism for cancer therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [PubMed]
- Keenan, M.M.; Chi, J.-T. Alternative fuels for cancer cells. Cancer J. 2015, 21, 49–55. [Google Scholar] [PubMed]
- Moffatt, B.A.; Ashihara, H. Purine and pyrimidine nucleotide synthesis and metabolism. Arab. Book/Am. Soc. Plant Biol. 2002, 1, e0018. [Google Scholar]
- Peng, H.; Wang, Y.; Luo, W. Multifaceted role of branched-chain amino acid metabolism in cancer. Oncogene 2020, 39, 6747–6756. [Google Scholar] [CrossRef]
- Green, C.R.; Wallace, M.; Divakaruni, A.S.; Phillips, S.A.; Murphy, A.N.; Ciaraldi, T.P.; Metallo, C.M. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol. 2016, 12, 15–21. [Google Scholar]
- Roberts, S.C.; Tancer, M.J.; Polinsky, M.R.; Gibson, K.M.; Heby, O.; Ullman, B. Arginase plays a pivotal role in polyamine precursor metabolism in Leishmania: Characterization of gene deletion mutants. J. Biol. Chem. 2004, 279, 23668–23678. [Google Scholar]
- Casero Jr, R.A.; Murray Stewart, T.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef]
- Sari, I.N.; Setiawan, T.; Kim, K.S.; Wijaya, Y.T.; Cho, K.W.; Kwon, H.Y. Metabolism and function of polyamines in cancer progression. Cancer Lett. 2021, 519, 91–104. [Google Scholar]
- Ha, H.C.; Yager, J.D.; Woster, P.A.; Casero Jr, R.A. Structural specificity of polyamines and polyamine analogues in the protection of DNA from strand breaks induced by reactive oxygen species. Biochem. Biophys. Res. Commun. 1998, 244, 298–303. [Google Scholar]
- Wu, G.; Meininger, C.J.; McNeal, C.J.; Bazer, F.W.; Rhoads, J.M. Role of L-Arginine in Nitric Oxide Synthesis and Health in Humans. Adv. Exp. Med. Biol. 2021, 1332, 167–187. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Oyaizu, T.; Enomoto, M.; Horie, M.; Yuasa, M.; Okawa, A.; Yagishita, K. VEGF and bFGF induction by nitric oxide is associated with hyperbaric oxygen-induced angiogenesis and muscle regeneration. Sci. Rep. 2020, 10, 2744. [Google Scholar] [CrossRef]
- Ridnour, L.A.; Isenberg, J.S.; Espey, M.G.; Thomas, D.D.; Roberts, D.D.; Wink, D.A. Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc. Natl. Acad. Sci. USA 2005, 102, 13147–13152. [Google Scholar]
- de Souza Galia, W.B.; Biazi, G.R.; Frasson-Uemura, I.G.; Miksza, D.R.; Zaia, C.T.B.V.; Zaia, D.A.M.; de Souza, H.M.; Bertolini, G.L. Gluconeogenesis is reduced from alanine, lactate and pyruvate, but maintained from glycerol, in liver perfusion of rats with early and late sepsis. Cell Biochem. Funct. 2021, 39, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Holeček, M. Origin and roles of Alanine and glutamine in Gluconeogenesis in the liver, kidneys, and small intestine under physiological and pathological conditions. Int. J. Mol. Sci. 2024, 25, 7037. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.; Gilewski, T.A.; Surbone, A.; Norton, L. Growth curve analysis. In Holland-Frei Cancer Medicine, 6th ed.; BC Decker: Hamilton, ON, Canada, 2003. [Google Scholar]
- Diana, A.; Carlino, F.; Franzese, E.; Oikonomidou, O.; Criscitiello, C.; De Vita, F.; Ciardiello, F.; Orditura, M. Early triple negative breast cancer: Conventional treatment and emerging therapeutic landscapes. Cancers 2020, 12, 819. [Google Scholar] [CrossRef]
- Costa, B.; Amorim, I.; Gärtner, F.; Vale, N. Understanding breast cancer: From conventional therapies to repurposed drugs. Eur. J. Pharm. Sci. 2020, 151, 105401. [Google Scholar]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef]
- Armstrong, J.S.; Whiteman, M.; Yang, H.; Jones, D.P.; Sternberg, P. Cysteine starvation activates the redox-dependent mitochondrial permeability transition in retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4183–4189. [Google Scholar] [CrossRef] [PubMed]
- Chiang, F.-F.; Chao, T.-H.; Huang, S.-C.; Cheng, C.-H.; Tseng, Y.-Y.; Huang, Y.-C. Cysteine regulates oxidative stress and glutathione-related antioxidative capacity before and after colorectal tumor resection. Int. J. Mol. Sci. 2022, 23, 9581. [Google Scholar] [CrossRef]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; De Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The interplay of reactive oxygen species, hypoxia, inflammation, and sirtuins in cancer initiation and progression. Oxid. Med. Cell Longev. 2016, 2016, 3907147. [Google Scholar] [CrossRef]
- Kung-Chun Chiu, D.; Pui-Wah Tse, A.; Law, C.-T.; Ming-Jing Xu, I.; Lee, D.; Chen, M.; Kit-Ho Lai, R.; Wai-Hin Yuen, V.; Wing-Sum Cheu, J.; Wai-Hung Ho, D. Hypoxia regulates the mitochondrial activity of hepatocellular carcinoma cells through HIF/HEY1/PINK1 pathway. Cell Death Dis. 2019, 10, 934. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Kostyuk, S.V.; Konkova, M.S.; Ershova, E.S.; Alekseeva, A.J.; Smirnova, T.D.; Stukalov, S.V.; Kozhina, E.A.; Shilova, N.V.; Zolotukhina, T.V.; Markova, Z.G.; et al. An exposure to the oxidized DNA enhances both instability of genome and survival in cancer cells. PLoS ONE 2013, 8, e77469. [Google Scholar] [CrossRef]
- Loseva, P.; Kostyuk, S.; Malinovskaya, E.; Clement, N.; Dechesne, C.; Dani, C.; Smirnova, T.; Glebova, K.; Baidakova, G.; Baranova, A. Extracellular DNA oxidation stimulates activation of NRF2 and reduces the production of ROS in human mesenchymal stem cells. Expert. Opin. Biol. Ther. 2012, 12, S85–S97. [Google Scholar] [CrossRef]
- Filev, A.D.; Shmarina, G.V.; Ershova, E.S.; Veiko, N.N.; Martynov, A.V.; Borzikova, M.A.; Poletkina, A.A.; Dolgikh, O.A.; Veiko, V.P.; Bekker, A.A.; et al. Oxidized Cell-Free DNA Role in the Antioxidant Defense Mechanisms under Stress. Oxid. Med. Cell Longev. 2019, 2019, 1245749. [Google Scholar] [CrossRef]
- Combs, J.A.; DeNicola, G.M. The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers 2019, 11, 678. [Google Scholar] [CrossRef]
- Tabata, S.; Endo, H.; Makinoshima, H.; Soga, T.; Inoue, M. The γ-glutamyl cycle serves as an amino acids supply system in colorectal cancer organoids under chronic hypoxia. Biochem. Biophys. Res. Commun. 2024, 714, 149977. [Google Scholar] [CrossRef]
- de Alteriis, E.; Incerti, G.; Cartenì, F.; Chiusano, M.L.; Colantuono, C.; Palomba, E.; Termolino, P.; Monticolo, F.; Esposito, A.; Bonanomi, G.; et al. Extracellular DNA secreted in yeast cultures is metabolism-specific and inhibits cell proliferation. Microb. Cell 2023, 10, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Palomba, E.; Chiusano, M.L.; Monticolo, F.; Langella, M.C.; Sanchez, M.; Tirelli, V.; de Alteriis, E.; Iannaccone, M.; Termolino, P.; Capparelli, R. Extracellular Self-DNA Effects on Yeast Cell Cycle and Transcriptome during Batch Growth. Biomolecules 2024, 14, 663. [Google Scholar] [CrossRef]
- Lindell, E.; Zhong, L.; Zhang, X. Quiescent Cancer Cells-A Potential Therapeutic Target to Overcome Tumor Resistance and Relapse. Int. J. Mol. Sci. 2023, 24, 3762. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, C.; Ling, S.; Wei, R.; Wang, J.; Xu, X. The metabolic flexibility of quiescent CSC: Implications for chemotherapy resistance. Cell Death Dis. 2021, 12, 835. [Google Scholar] [CrossRef]
- Truskowski, K.; Amend, S.R.; Pienta, K.J. Dormant cancer cells: Programmed quiescence, senescence, or both? Cancer Metastasis Rev. 2023, 42, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Steinbichler, T.B.; Savic, D.; Dudás, J.; Kvitsaridze, I.; Skvortsov, S.; Riechelmann, H.; Skvortsova, I.I. Cancer stem cells and their unique role in metastatic spread. Semin. Cancer Biol. 2020, 60, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Michelatti, D.; Beyes, S.; Bernardis, C.; Negri, M.L.; Morelli, L.; Bediaga, N.G.; Poli, V.; Fagnocchi, L.; Lago, S.; D′Annunzio, S.; et al. Oncogenic enhancers prime quiescent metastatic cells to escape NK immune surveillance by eliciting transcriptional memory. Nat. Commun. 2024, 15, 2198. [Google Scholar] [CrossRef]
- De Donatis, A.; Ranaldi, F.; Cirri, P. Reciprocal control of cell proliferation and migration. Cell Commun. Signal. 2010, 8, 20. [Google Scholar] [CrossRef]
- Tlili, S.; Gauquelin, E.; Li, B.; Cardoso, O.; Ladoux, B.; Delanoë-Ayari, H.; Graner, F. Collective cell migration without proliferation: Density determines cell velocity and wave velocity. R. Soc. Open Sci. 2018, 5, 172421. [Google Scholar]
- Zanca, A.; Flegg, J.A.; Osborne, J.M. Push or pull? Cell proliferation and migration during wound healing. Front. Syst. Biol. 2022, 2, 876075. [Google Scholar]
- Alfahed, A. Cell Migration–Proliferation Dichotomy in Cancer: Biological Fact or Experimental Artefact? Biology 2024, 13, 753. [Google Scholar] [CrossRef]
- de Alteriis, E.; Cartenì, F.; Parascandola, P.; Serpa, J.; Mazzoleni, S. Revisiting the Crabtree/Warburg effect in a dynamic perspective: A fitness advantage against sugar-induced cell death. Cell Cycle 2018, 17, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Park, B.; Brinkmann, M.M.; Spooner, E.; Lee, C.C.; Kim, Y.M.; Ploegh, H.L. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat. Immunol. 2008, 9, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.S.; Cameron, J.; Brooks, J.C.; Leifer, C.A. Complex negative regulation of TLR9 by multiple proteolytic cleavage events. J. Immunol. 2016, 197, 1343–1352. [Google Scholar] [PubMed]
- Chockalingam, A.; Brooks, J.C.; Cameron, J.L.; Blum, L.K.; Leifer, C.A. TLR9 traffics through the Golgi complex to localize to endolysosomes and respond to CpG DNA. Immunol. Cell Biol. 2009, 87, 209–217. [Google Scholar] [CrossRef]
- Combes, A.; Camosseto, V.; N’guessan, P.; Argüello, R.J.; Mussard, J.; Caux, C.; Bendriss-Vermare, N.; Pierre, P.; Gatti, E. BAD-LAMP controls TLR9 trafficking and signalling in human plasmacytoid dendritic cells. Nat. Commun. 2017, 8, 913. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lemos, I.; Freitas-Dias, C.; Hipólito, A.; Ramalho, J.; Carteni, F.; Gonçalves, L.G.; Mazzoleni, S.; Serpa, J. Cell-Free DNA (cfDNA) Regulates Metabolic Remodeling, Sustaining Proliferation, Quiescence, and Migration in MDA-MB-231, a Triple-Negative Breast Carcinoma (TNBC) Cell Line. Metabolites 2025, 15, 227. https://doi.org/10.3390/metabo15040227
Lemos I, Freitas-Dias C, Hipólito A, Ramalho J, Carteni F, Gonçalves LG, Mazzoleni S, Serpa J. Cell-Free DNA (cfDNA) Regulates Metabolic Remodeling, Sustaining Proliferation, Quiescence, and Migration in MDA-MB-231, a Triple-Negative Breast Carcinoma (TNBC) Cell Line. Metabolites. 2025; 15(4):227. https://doi.org/10.3390/metabo15040227
Chicago/Turabian StyleLemos, Isabel, Catarina Freitas-Dias, Ana Hipólito, José Ramalho, Fabrizio Carteni, Luís G. Gonçalves, Stefano Mazzoleni, and Jacinta Serpa. 2025. "Cell-Free DNA (cfDNA) Regulates Metabolic Remodeling, Sustaining Proliferation, Quiescence, and Migration in MDA-MB-231, a Triple-Negative Breast Carcinoma (TNBC) Cell Line" Metabolites 15, no. 4: 227. https://doi.org/10.3390/metabo15040227
APA StyleLemos, I., Freitas-Dias, C., Hipólito, A., Ramalho, J., Carteni, F., Gonçalves, L. G., Mazzoleni, S., & Serpa, J. (2025). Cell-Free DNA (cfDNA) Regulates Metabolic Remodeling, Sustaining Proliferation, Quiescence, and Migration in MDA-MB-231, a Triple-Negative Breast Carcinoma (TNBC) Cell Line. Metabolites, 15(4), 227. https://doi.org/10.3390/metabo15040227