

A Multifaceted Computational Approach to Identify PAD4 Inhibitors for the Treatment of Rheumatoid Arthritis (RA)

 , , , , , ,
, , , , , ,
Abstract

1. Introduction
2. Materials and Methods
2.1. Computational Tools
2.2. Generation of Databases and Ligand Library Preparation
2.3. Crystal Structure Retrieval and Preparation
2.4. Binding Pocket Determination and Validation of Molecular Docking
2.5. Standard Molecular Docking (Rigid)
2.6. Induced-Fit Docking (IFD) (Flexible)
2.7. Molecular Mechanics-Based Re-Scoring
−(ΔG solvation, ligand + ΔG solvation, receptor).
2.8. Shape-Based Screen
2.9. Quantum Chemical Calculations
3. Results and Discussion
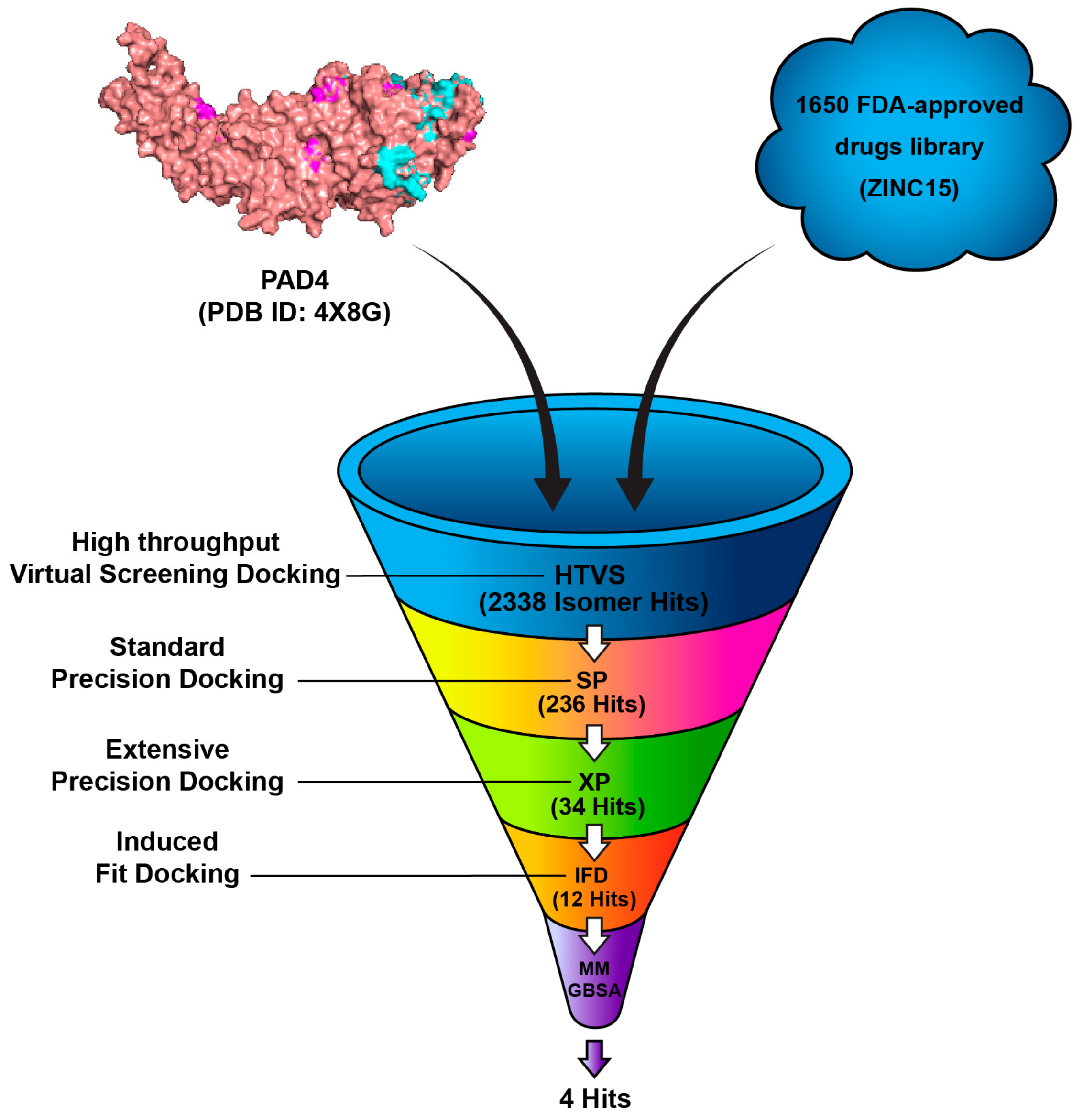
3.1. Docking Studies
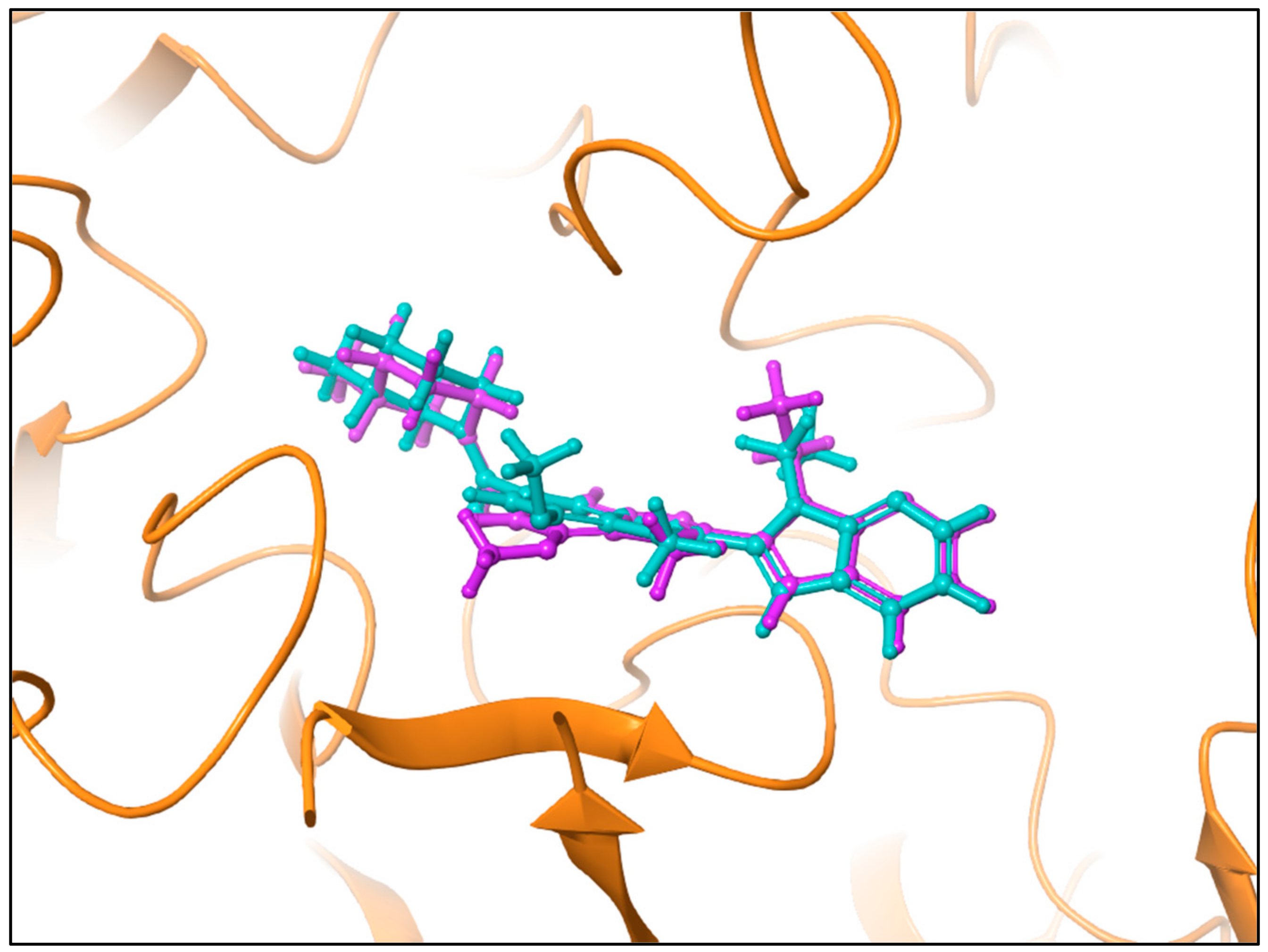
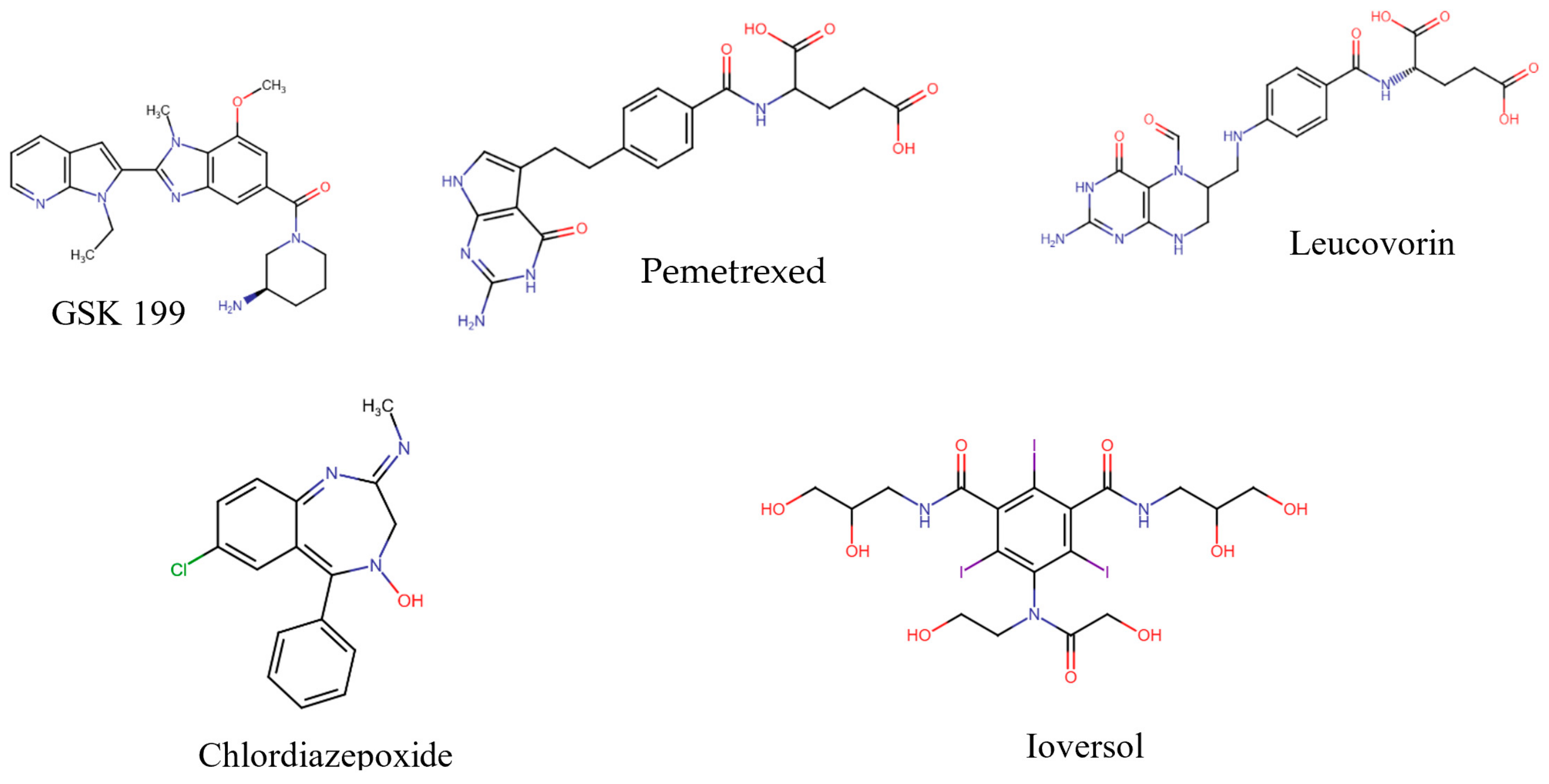
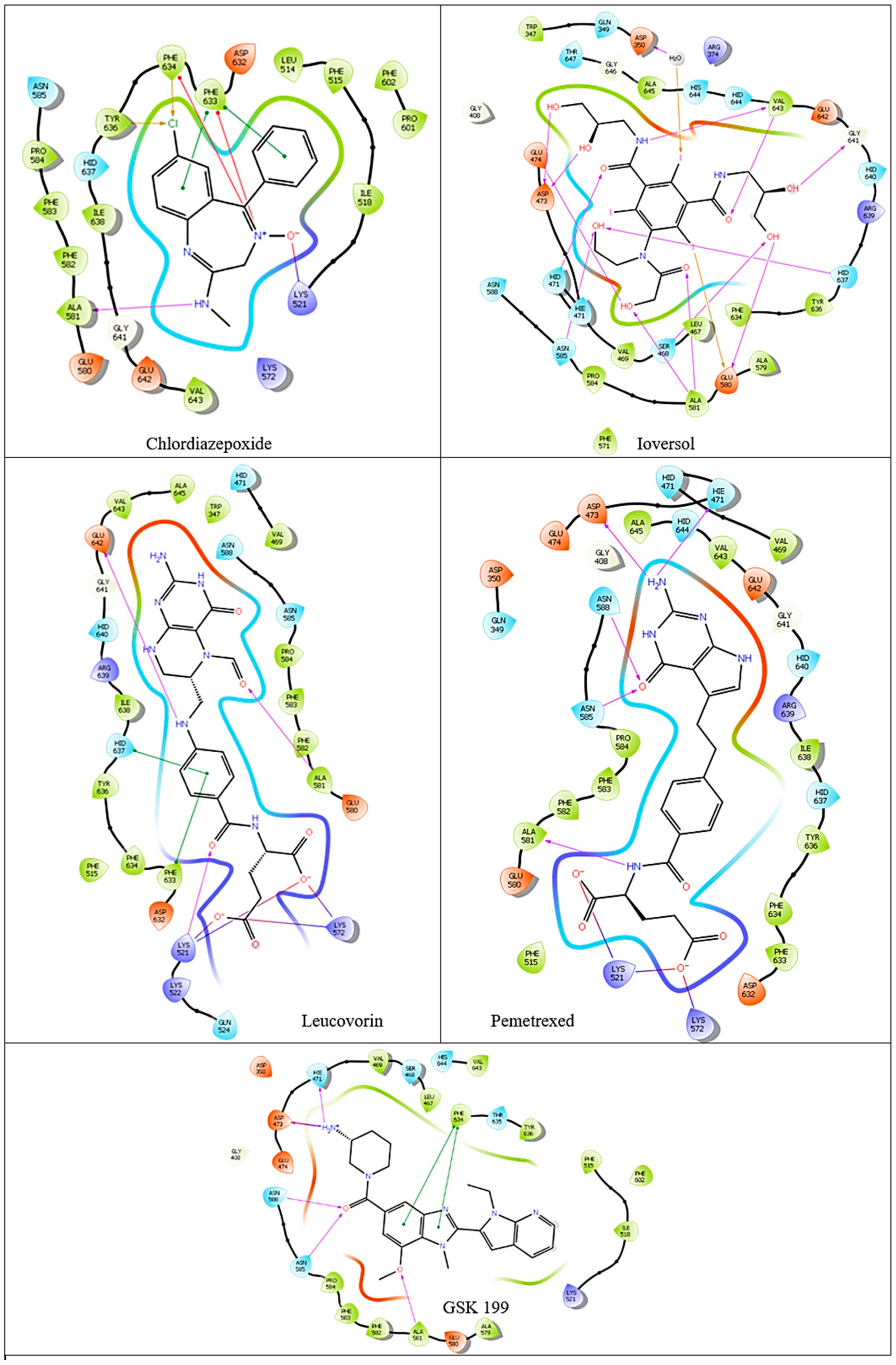
3.2. Computational Analysis of the Four Hits Binding to PAD4
3.3. Binding Free Energies Analysis
3.4. Shape Similarity Prediction
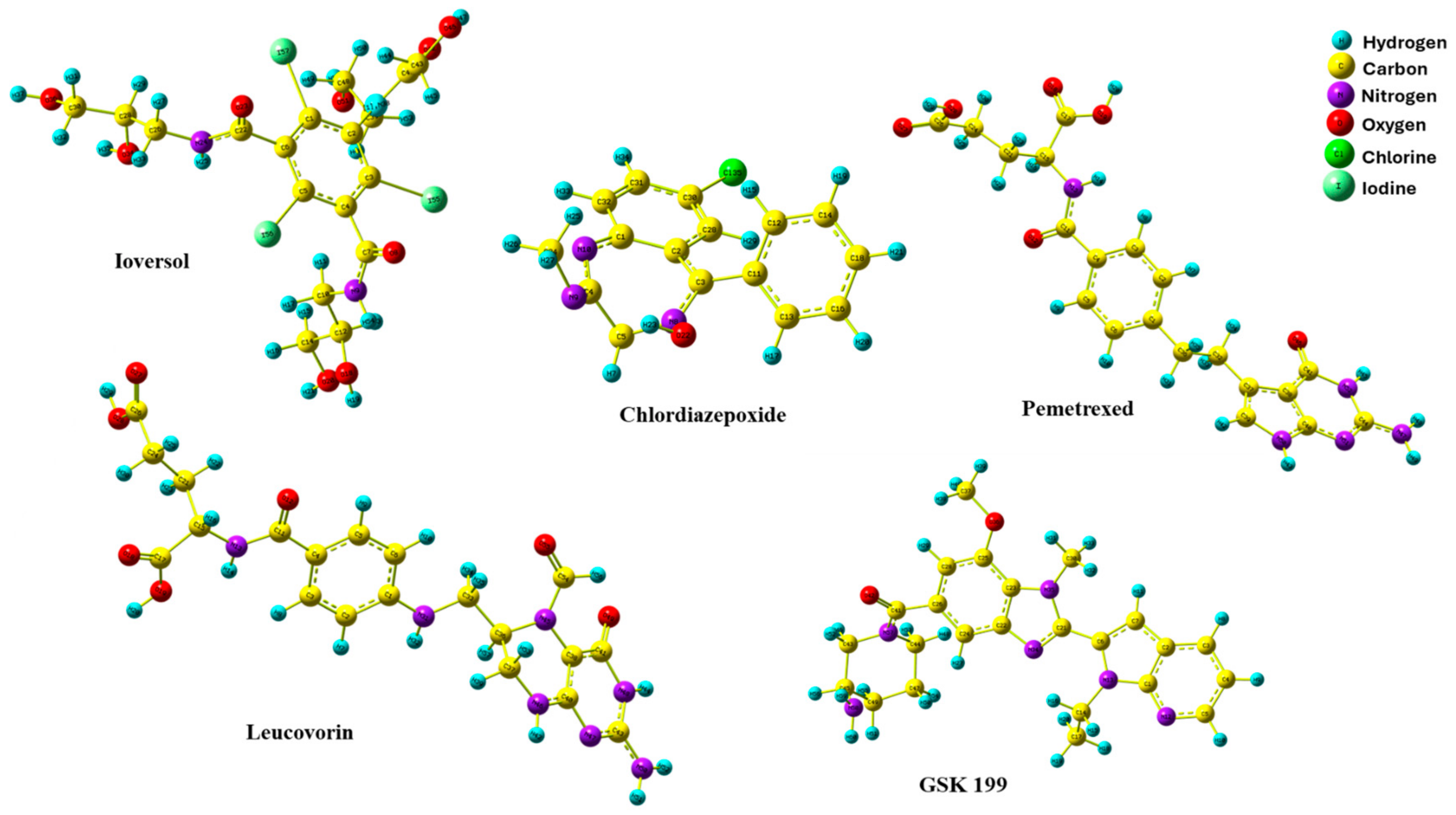
3.5. DFT Optimization Structures
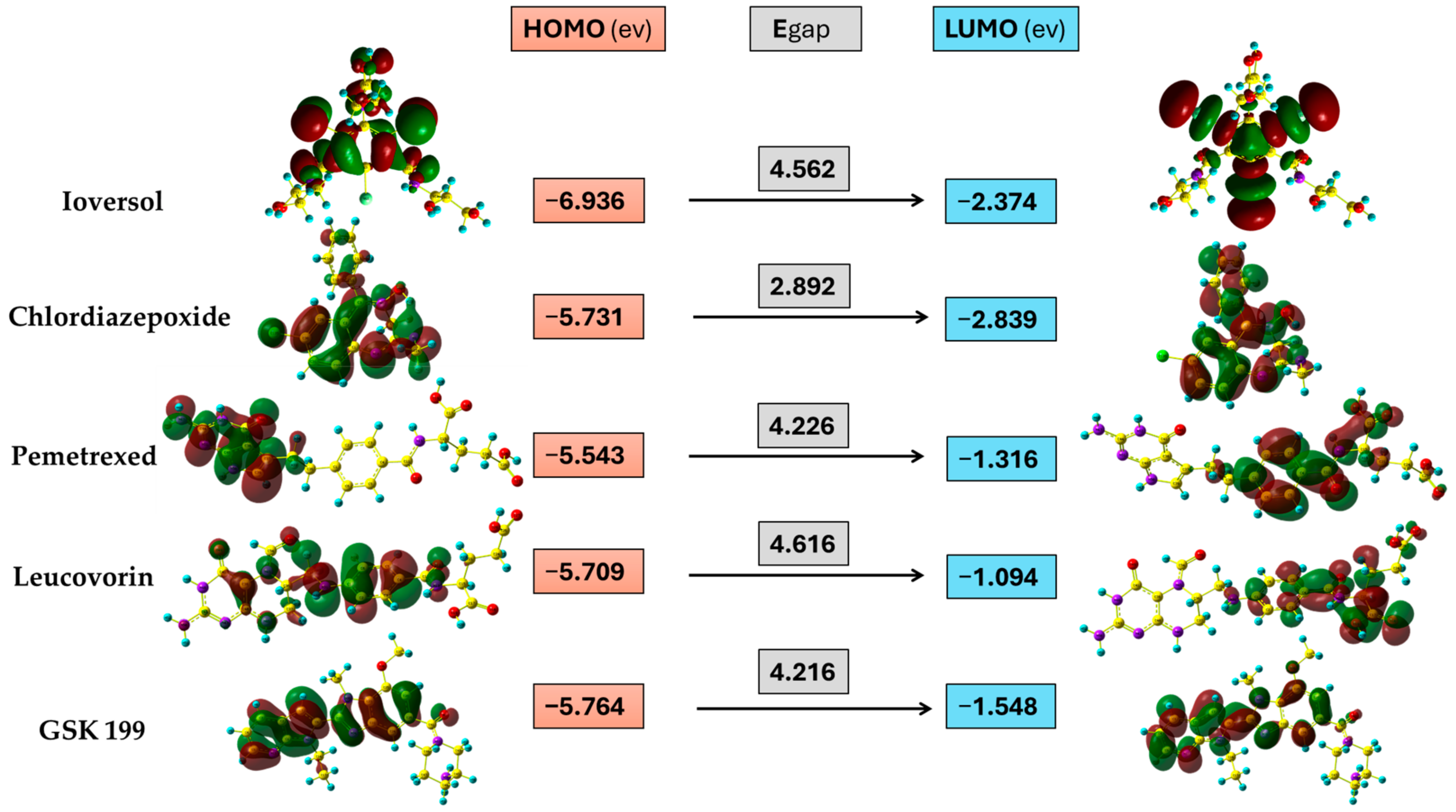
3.6. Frontier Molecular Orbital
3.7. Global Chemical Descriptors
3.8. Molecular Electrostatic Potential and Mulliken Population Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vossenaar, E.R.; Zendman, A.J.W.; Van Venrooij, W.J.; Pruijn, G.J.M. PAD, a Growing Family of Citrullinating Enzymes: Genes, Features and Involvement in Disease. BioEssays 2003, 25, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.E.; Causey, C.P.; Knuckley, B.; Slack-Noyes, J.L.; Thompson, P.R. Protein Arginine Deiminase 4 (PAD4): Current Understanding and Future Therapeutic Potential. Curr. Opin. Drug Discov. Dev. 2009, 12, 616–627. [Google Scholar] [PubMed] [PubMed Central]
- Witalison, E.E.; Thompson, P.R.; Hofseth, L.J. Protein arginine deiminases and associated citrullination: Physiological functions and diseases associated with dysregulation. Curr. Drug Targets 2015, 16, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Asaga, H.; Yamada, M.; Senshu, T. Selective deimination of vimentin in calcium ionophore-induced apoptosis of mouse peritoneal macrophages. Biochem. Biophys. Res. Commun. 1998, 243, 641–646. [Google Scholar] [CrossRef]
- Koushik, S.; Joshi, N.; Nagaraju, S.; Mahmood, S.; Mudeenahally, K.; Padmavathy, R.; Jegatheesan, S.K.; Mullangi, R.; Rajagopal, S. PAD4: Pathophysiology, current therapeutics and future perspective in rheumatoid arthritis. Expert Opin. Ther. Targets 2017, 21, 433–447. [Google Scholar] [CrossRef]
- Sabnis, R.W. Novel Peptidylarginine Deiminase Type 4 (PAD4) Inhibitors. ACS Med. Chem. Lett. 2022, 13, 1537–1538. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Zee, B.M.; Levin, R.S.; DiMaggio, P.A.; Garcia, B.A. Global turnover of histone post-translational modifications and variants in human cells. Epigenetics Chromatin 2010, 3, 22. [Google Scholar] [CrossRef]
- Bicker, K.L.; Thompson, P.R. The protein arginine deiminases: Structure, function, inhibition, and disease. Biopolymers 2013, 99, 155–163. [Google Scholar] [CrossRef]
- Arita, K.; Hashimoto, H.; Shimizu, T.; Nakashima, K.; Yamada, M.; Sato, M. Structural basis for Ca2+-induced activation of human PAD4. Nat. Struct. Mol. Biol. 2004, 11, 777–783. [Google Scholar] [CrossRef]
- Cherrington, B.D.; Morency, E.; Struble, A.M.; Coonrod, S.A.; Wakshlag, J.J. Potential role for peptidylarginine deiminase 2 (PAD2) in citrullination of canine mammary epithelial cell histones. PLoS ONE 2010, 5, e11768. [Google Scholar] [CrossRef]
- Jang, B.; Shin, H.Y.; Choi, J.K.; Nguyen, D.P.T.; Jeong, B.H.; Ishigami, A.; Maruyama, N.; Carp, R.I.; Kim, Y.S.; Choi, E.K. Subcellular localization of peptidylarginine deiminase 2 and citrullinated proteins in brains of scrapie-infected mice: Nuclear localization of PAD2 and membrane fraction-enriched citrullinated proteins. J. Neuropathol. Exp. Neurol. 2011, 70, 116–124. [Google Scholar] [CrossRef]
- Darrah, E.; Rosen, A.; Giles, J.T.; Andrade, F. Peptidylarginine deiminase 2, 3 and 4 have distinct specificities against cellular substrates: Novel insights into autoantigen selection in rheumatoid arthritis. Ann. Rheum. Dis. 2012, 71, 92–98. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Masson-Bessière, C.; Sebbag, M.; Girbal-Neuhauser, E.; Nogueira, L.; Vincent, C.; Senshu, T.; Serre, G. The major synovial targets of the rheumatoid arthritis-specific antifilaggrin autoantibodies are deiminated forms of the α-and β-chains of fibrin. J. Immunol. 2001, 166, 4177–4184. [Google Scholar] [CrossRef]
- Yu, K.; Proost, P. Insights into peptidylarginine deiminase expression and citrullination pathways. Trends Cell Biol. 2022, 32, 746–761. [Google Scholar] [CrossRef]
- Curran, A.M.; Naik, P.; Giles, J.T.; Darrah, E. PAD enzymes in rheumatoid arthritis: Pathogenic effectors and autoimmune targets. Nat. Rev. Rheumatol. 2020, 16, 301–315. [Google Scholar] [CrossRef]
- Yang, C.; Dong, Z.Z.; Zhang, J.; Teng, D.; Luo, X.; Li, D.; Zhou, Y. Peptidylarginine deiminases 4 as a promising target in drug discovery. Eur. J. Med. Chem. 2021, 226, 113840. [Google Scholar] [CrossRef]
- Martinez-Prat, L.; Palterer, B.; Vitiello, G.; Parronchi, P.; Robinson, W.H.; Mahler, M. Autoantibodies to protein-arginine deiminase (PAD) 4 in rheumatoid arthritis: Immunological and clinical significance, and potential for precision medicine: Anti-PAD4 antibodies in RA. Expert Rev. Clin. Immunol. 2019, 15, 1073–1087. [Google Scholar] [CrossRef]
- Damgaard, D.; Bawadekar, M.; Senolt, L.; Stensballe, A.; Shelef, M.A.; Nielsen, C.H. Relative efficiencies of peptidylarginine deiminase 2 and 4 in generating target sites for anti-citrullinated protein antibodies in fibrinogen, alpha-enolase and histone H3. PLoS ONE 2018, 13, e0203214. [Google Scholar] [CrossRef]
- Martín Monreal, M.T.; Rebak, A.S.; Massarenti, L.; Mondal, S.; Šenolt, L.; Ødum, N.; Nielsen, M.L.; Thompson, P.R.; Nielsen, C.H.; Damgaard, D. Applicability of small-molecule inhibitors in the study of peptidyl arginine deiminase 2 (PAD2) and PAD4. Front. Immunol. 2021, 12, 716250. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.; Lin, J. Theoretical study of the mechanism of protein arginine deiminase 4 (PAD4) inhibition by F-amidine. J. Mol. Graph. Model. 2015, 55, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Arita, K.; Bhatia, M.; Knuckley, B.; Lee, Y.H.; Stallcup, M.R.; Sato, M.; Thompson, P.R. Inhibitors and inactivators of protein arginine deiminase 4: Functional and structural characterization. Biochemistry 2006, 45, 11727–11736. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Luo, Y.; Knuckley, B.; Lee, Y.H.; Stallcup, M.R.; Thompson, P.R. A fluoroacetamidine-based inactivator of protein arginine deiminase 4: Design, synthesis, and in vitro and in vivo evaluation. J. Am. Chem. Soc. 2006, 128, 1092–1093. [Google Scholar] [CrossRef]
- Lewis, H.D.; Liddle, J.; Coote, J.E.; Atkinson, S.J.; Barker, M.D.; Bax, B.D.; Bicker, K.L.; Bingham, R.P.; Campbell, M.; Chen, Y.H.; et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 2015, 11, 189–191. [Google Scholar] [CrossRef]
- Jones, J.E.; Slack, J.L.; Fang, P.; Zhang, X.; Subramanian, V.; Causey, C.P.; Coonrod, S.A.; Guo, M.; Thompson, P.R. Synthesis and screening of a haloacetamidine containing library to identify PAD4 selective inhibitors. ACS Chem. Biol. 2012, 7, 160–165. [Google Scholar] [CrossRef]
- Causey, C.P.; Jones, J.E.; Slack, J.L.; Kamei, D.; Jones Jr, L.E.; Subramanian, V.; Knuckley, B.; Ebrahimi, P.; Chumanevich, A.A.; Luo, Y.; et al. The development of N-α-(2-carboxyl) benzoyl-N 5-(2-fluoro-1-iminoethyl)-l-ornithine amide (o-F-amidine) and N-α-(2-carboxyl) benzoyl-N 5-(2-chloro-1-iminoethyl)-l-ornithine amide (o-Cl-amidine). As second-generation protein arginine deiminase (PAD) inhibitors. J. Med. Chem. 2011, 54, 6919–6935. [Google Scholar] [CrossRef]
- Knuckley, B.; Luo, Y.; Thompson, P.R. Profiling Protein Arginine Deiminase 4 (PAD4): A novel screen to identify PAD4 inhibitors. Bioorganic Med. Chem. 2008, 16, 739–745. [Google Scholar] [CrossRef]
- Osada, A.; Matsumoto, I.; Mikami, N.; Ohyama, A.; Kurata, I.; Kondo, Y.; Tsuboi, H.; Ishigami, A.; Sano, Y.; Arai, T.; et al. Citrullinated inter-alpha-trypsin inhibitor heavy chain 4 in arthritic joints and its potential effect in the neutrophil migration. Clin. Exp. Immunol. 2021, 203, 385–399. [Google Scholar] [CrossRef]
- Ali, S.; Nazir, A.; Shabbir, S.; Ashraf, S. Understanding the role of peptidylarginine deiminases (PADs) in diseases and their inhibitors as potential therapeutic agents. Eur. J. Pharm. Med. Res. 2017, 4, 184–192. [Google Scholar]
- Ahmad, F.; Albutti, A.; Tariq, M.H.; Din, G.; Tahir ul Qamar, M.; Ahmad, S. Discovery of potential antiviral compounds against hendra virus by targeting its receptor-binding protein (G) using computational approaches. Molecules 2022, 27, 554. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Jaundoo, R.; Bohmann, J.; Gutierrez, G.E.; Klimas, N.; Broderick, G.; Craddock, T.J. Using a consensus docking approach to predict adverse drug reactions in combination drug therapies for gulf war illness. Int. J. Mol. Sci. 2018, 19, 3355. [Google Scholar] [CrossRef]
- Aldholmi, M.; Ahmad, R.; Shaikh, M.H.; Salem, A.M.; Alqurashi, M.; Alturki, M. Anti-Infective Activity of Momordica charantia Extract with Molecular Docking of Its Triterpenoid Glycosides. Antibiotics 2024, 13, 544. [Google Scholar] [CrossRef]
- Alturki, M.S. Exploring marine-derived compounds: In silico discovery of selective ketohexokinase (KHK) inhibitors for metabolic disease therapy. Mar. Drugs 2024, 22, 455. [Google Scholar] [CrossRef]
- Rants’O, T.A.; van der Westhuizen, C.J.; van Zyl, R.L. Optimization of covalent docking for organophosphates interaction with Anopheles acetylcholinesterase. J. Mol. Graph. Model. 2022, 110, 108054. [Google Scholar] [CrossRef]
- Dehbanipour, R.; Ghalavand, Z. Acinetobacter baumannii: Pathogenesis, virulence factors, novel therapeutic options and mechanisms of resistance to antimicrobial agents with emphasis on tigecycline. J. Clin. Pharm. Ther. 2022, 47, 1875–1884. [Google Scholar] [CrossRef]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Vila, J.; Pachon, J. Therapeutic options for Acinetobacter baumannii infections: An update. Expert Opin. Pharmacother. 2012, 13, 2319–2336. [Google Scholar] [CrossRef]
- Potron, A.; Poirel, L.; Nordmann, P. Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: Mechanisms and epidemiology. Int. J. Antimicrob. Agents 2015, 45, 568–585. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, K.Y.J. Advances in the development of shape similarity methods and their application in drug discovery. Front. Chem. 2018, 6, 315. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Hay, P.J.; Willard, R.W. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Domingo, L.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Roy, D.R. Update 1 of: Electrophilicity index. Chem. Rev. 2007, 107, PR46–PR74. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, A.F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Maiti, B. HSAB principle applied to the time evolution of chemical reactions. J. Am. Chem. Soc. 2003, 125, 2705–2710. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Natl. Acad. Sci. USA 1986, 83, 8440–8441. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute electronegativity and hardness: Applications to organic chemistry. J. Org. Chem. 1989, 54, 1423–1430. [Google Scholar] [CrossRef]
- Fukui, K.; Yonezawa, T.; Nagata, C.; Shingu, H. Molecular orbital theory of orientation in aromatic, heteroaromatic, and other conjugated molecules. J. Chem. Phys. 1954, 22, 1433–1442. [Google Scholar] [CrossRef]
- Ruiz-Morales, Y. HOMO−LUMO gap as an index of molecular size and structure for polycyclic aromatic hydrocarbons (PAHs) and asphaltenes: A theoretical study. I. J. Phys. Chem. A 2002, 106, 11283–11308. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. Statistical analysis of the molecular surface electrostatic potential: An approach to describing noncovalent interactions in condensed phases. J. Mol. Struct. THEOCHEM 1998, 425, 107–114. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Sastry, G.M.; Dixon, S.L.; Sherman, W. Rapid shape-based ligand alignment and virtual screening method based on atom/feature-pair similarities and volume overlap scoring. J. Chem. Inf. Model. 2011, 51, 2455–2466. [Google Scholar] [CrossRef]
- Rosales-López, A.; López-Castillo, G.N.; Sandoval-Ramírez, J.; Terán, J.L.; Carrasco-Carballo, A. Correlation between Molecular Docking and the Stabilizing Interaction of HOMO-LUMO: Spirostans in CHK1 and CHK2, an In Silico Cancer Approach. Int. J. Mol. Sci. 2024, 25, 8588. [Google Scholar] [CrossRef]
- Gheidari, D.; Morteza, M.; Foroozan, H. Virtual screening, molecular docking, MD simulation studies, DFT calculations, ADMET, and drug likeness of Diaza-adamantane as potential MAPKERK inhibitors. Front. Pharmacol. 2024, 15, 13. [Google Scholar] [CrossRef]
- Nehra, N.; Tittal, R.K.; Ghule, V.D. 1, 2, 3-Triazoles of 8-hydroxyquinoline and HBT: Synthesis and studies (DNA binding, antimicrobial, molecular docking, ADME, and DFT). ACS Omega 2021, 6, 27089–27100. [Google Scholar] [CrossRef] [PubMed]
- Al Sheikh Ali, A.; Khan, D.; Naqvi, A.; Al-Blewi, F.F.; Rezki, N.; Aouad, M.R.; Hagar, M. Design, synthesis, molecular modeling, anticancer studies, and density functional theory calculations of 4-(1, 2, 4-triazol-3-ylsulfanylmethyl)-1, 2, 3-triazole derivatives. ACS Omega 2020, 6, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Vargas, R.; Garza, J.; Cedillo, A. Koopmans-like approximation in the Kohn− Sham method and the impact of the frozen core approximation on the computation of the reactivity parameters of the density functional theory. J. Phys. Chem. A 2005, 109, 8880–8892. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Irreversible amidines (Pan inhibitors) | IC50 on PAD4 |
|---|---|
| F-amidine | 1.9 µM |
| Cl-amidine | 22 µM |
| Reversible compounds (Selective) | |
| GSK199 | 200 nM |
| GSK484 | 50 nM |
| Compound | Glide Score Docking (Rigid) | Induced-Fit Docking (IFD) (Flexible) | Ionic Interactions | H-Bond Interactions | Pi Pi-Bond Interactions |
|---|---|---|---|---|---|
| Ioversol | −8.35 | −11.617 | - | HID471 ASP473 GLU474 GLU580 ALA581 GLY641 H2O350 | - |
| Pemetrexed | −8.28 | −10.599 | LYS521 LYS572 | HIE471 ASP473 ASN585 ASN588 ALA581 | - |
| Chlordiazepoxide | −5.23 | −9.988 | LYS521 PHE633 PHE634 | ALA581 | PHE633 |
| Leucovorin | −4.16 | −10.521 | LYS521 LYS572 | LYS521 ALA581 GLU642 | PHE633 HID637 |
| GSK199 | −9.58 | - | HIE471 ASP473 | ALA581 ASN585 ASN588 | PHE634 |
| Compound | ΔG binding a |
|---|---|
| Ioversol | −53.53 |
| Leucovorin | −43.71 |
| Chlordiazepoxide | −30.96 |
| Pemetrexed | −28.46 |
| GSK199 (Control) | −107.15 |
| Compound | Shape Similarity a |
|---|---|
| Ioversol | 0.212 |
| Leucovorin | 0.258 |
| Chlordiazepoxide | 0.280 |
| Pemetrexed | 0.300 |
| GSK199 | 1 |
| Compound | HOMO | LUMO | Global Hardness (η) | Global Softness (σ) | Electronegativity (χ) | Electrophilicity Index (ώ) |
|---|---|---|---|---|---|---|
| Ioversol | −6.936 | −2.374 | 2.281 | 0.438 | 4.655 | 5.934 |
| Pemetrexed | −5.543 | −1.316 | 2.113 | 0.473 | 3.430 | 4.718 |
| Chlordiazepoxide | −5.731 | −2.839 | 1.446 | 0.691 | 4.285 | 1.512 |
| Leucovorin | −5.709 | −1.094 | 2.308 | 0.433 | 3.402 | 6.147 |
| GSK199 | −5.764 | −1.548 | 2.108 | 0.474 | 3.656 | 4.685 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alturki, M.S.; Gomaa, M.S.; Tawfeeq, N.; Al Khzem, A.H.; Shaik, M.B.; Alshaikh Jafar, M.; Alsamen, M.; Al Nahab, H.; Al-Eid, M.; Almutawah, A.; et al. A Multifaceted Computational Approach to Identify PAD4 Inhibitors for the Treatment of Rheumatoid Arthritis (RA). Metabolites 2025, 15, 156. https://doi.org/10.3390/metabo15030156
Alturki MS, Gomaa MS, Tawfeeq N, Al Khzem AH, Shaik MB, Alshaikh Jafar M, Alsamen M, Al Nahab H, Al-Eid M, Almutawah A, et al. A Multifaceted Computational Approach to Identify PAD4 Inhibitors for the Treatment of Rheumatoid Arthritis (RA). Metabolites. 2025; 15(3):156. https://doi.org/10.3390/metabo15030156
Chicago/Turabian StyleAlturki, Mansour S., Mohamed S. Gomaa, Nada Tawfeeq, Abdulaziz H. Al Khzem, Mohsina B. Shaik, Murtadha Alshaikh Jafar, Mohammad Alsamen, Hasan Al Nahab, Mohammad Al-Eid, Alhassan Almutawah, and et al. 2025. "A Multifaceted Computational Approach to Identify PAD4 Inhibitors for the Treatment of Rheumatoid Arthritis (RA)" Metabolites 15, no. 3: 156. https://doi.org/10.3390/metabo15030156
APA StyleAlturki, M. S., Gomaa, M. S., Tawfeeq, N., Al Khzem, A. H., Shaik, M. B., Alshaikh Jafar, M., Alsamen, M., Al Nahab, H., Al-Eid, M., Almutawah, A., Rants’o, T. A., Ayil, K. A. G., & Almaghrabi, M. (2025). A Multifaceted Computational Approach to Identify PAD4 Inhibitors for the Treatment of Rheumatoid Arthritis (RA). Metabolites, 15(3), 156. https://doi.org/10.3390/metabo15030156