
Nicotinamide N-Methyltransferase (NNMT): A New Hope for Treating Aging and Age-Related Conditions


Abstract
1. Introduction
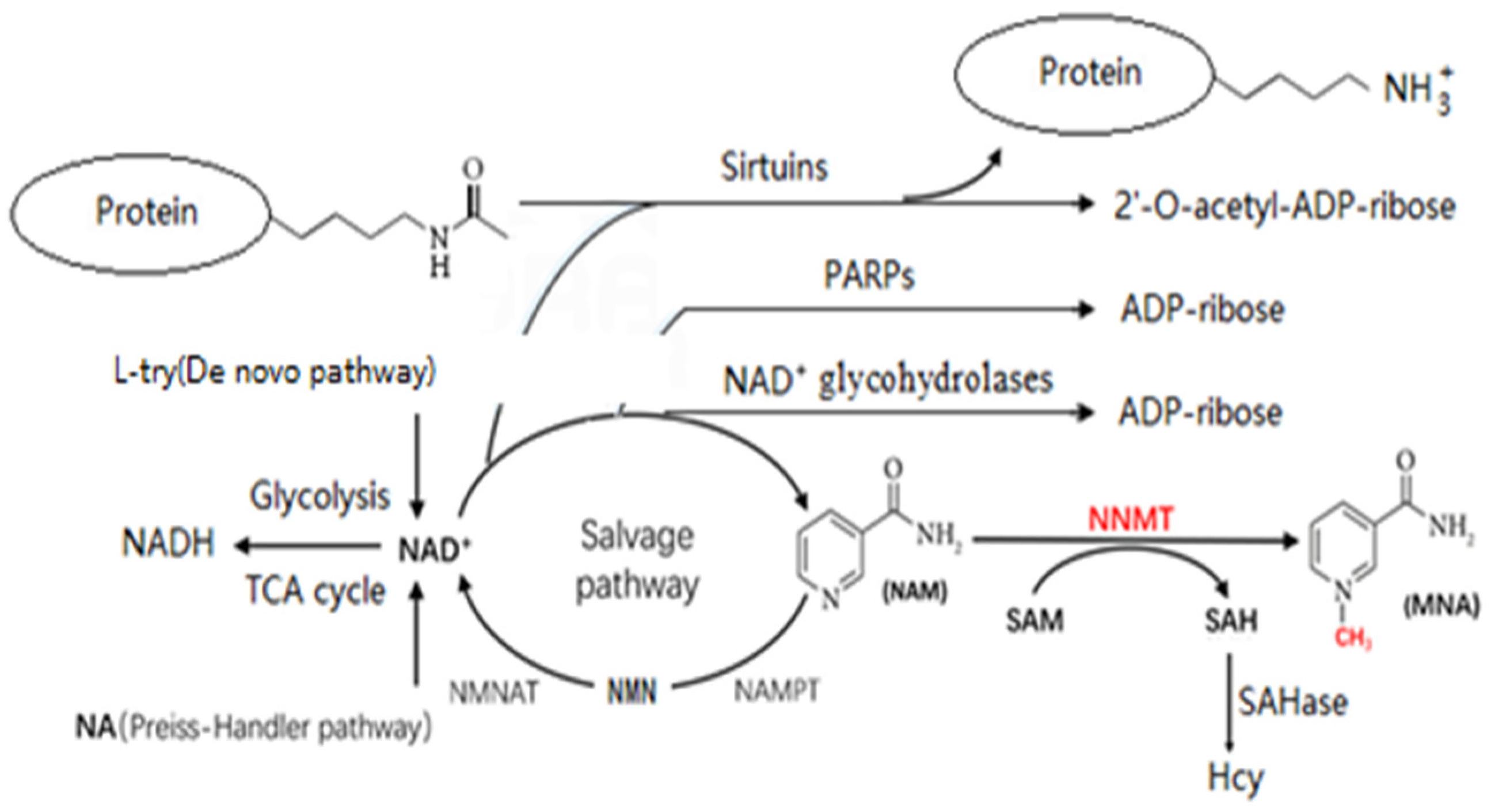
2. Potential Metabolic Pathways Linking NNMT to Aging
2.1. Consequences of Increased Homocysteine Levels
2.2. Consequences of Decreasing NAD+ Levels
3. Possible Mechanisms by Which NNMT Affects Aging through Hcy
3.1. Hcy and Mitochondrial Dysfunction
3.2. Hcy and Cellular Aging
3.3. Hcy and Oxidative Stress
3.4. Hcy and Inflammation
4. Possible Mechanisms by Which NNMT Affects Aging through NAD+
4.1. Decreased NAD+ Levels and Aging
4.2. Sirtuins and Aging
4.3. PARPs and Aging
4.4. NAD+ Glycohydrolases and Aging
5. Research Progress on NNMT in Aging-Related Diseases
5.1. NNMT and Cancer
5.2. NNMT and Diabetes
5.3. NNMT and Cardiovascular Diseases
5.4. NNMT and Neurodegenerative Diseases
6. NNMT as a Therapeutic Target
7. Prospects for Future Research
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Martínez, P.; Blasco, M.A. Heart-Breaking Telomeres. Circ. Res. 2018, 123, 787–802. [Google Scholar] [CrossRef]
- Aksoy, S.; Szumlanski, C.L.; Weinshilboum, R.M. Human Liver Nicotinamide N-Methyltransferase. cDNA Cloning, Expression, and Biochemical Characterization. J. Biol. Chem. 1994, 269, 14835–14840. [Google Scholar] [CrossRef]
- Alston, T.A.; Abeles, R.H. Substrate Specificity of Nicotinamide Methyltransferase Isolated from Porcine Liver. Arch. Biochem. Biophys. 1988, 260, 601–608. [Google Scholar] [CrossRef]
- Bockwoldt, M.; Houry, D.; Niere, M.; Gossmann, T.I.; Reinartz, I.; Schug, A.; Ziegler, M.; Heiland, I. Identification of Evolutionary and Kinetic Drivers of NAD-Dependent Signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 15957–15966. [Google Scholar] [CrossRef]
- Komatsu, M.; Kanda, T.; Urai, H.; Kurokochi, A.; Kitahama, R.; Shigaki, S.; Ono, T.; Yukioka, H.; Hasegawa, K.; Tokuyama, H.; et al. NNMT Activation Can Contribute to the Development of Fatty Liver Disease by Modulating the NAD + Metabolism. Sci. Rep. 2018, 8, 8637. [Google Scholar] [CrossRef]
- Pissios, P. Nicotinamide N-Methyltransferase: More Than a Vitamin B3 Clearance Enzyme. Trends Endocrinol. Metab. 2017, 28, 340–353. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Kale, A.; Perrone, R.; Lopez-Dominguez, J.A.; Pisco, A.O.; Kasler, H.G.; Schmidt, M.S.; Heckenbach, I.; Kwok, R.; Wiley, C.D.; et al. Senescent Cells Promote Tissue NAD+ Decline during Ageing via the Activation of CD38+ Macrophages. Nat. Metab. 2020, 2, 1265–1283. [Google Scholar] [CrossRef]
- Gokarn, R.; Solon-Biet, S.M.; Cogger, V.C.; Cooney, G.J.; Wahl, D.; McMahon, A.C.; Mitchell, J.R.; Mitchell, S.J.; Hine, C.; de Cabo, R.; et al. Long-Term Dietary Macronutrients and Hepatic Gene Expression in Aging Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1618–1625. [Google Scholar] [CrossRef]
- Neelakantan, H.; Brightwell, C.R.; Graber, T.G.; Maroto, R.; Wang, H.-Y.L.; McHardy, S.F.; Papaconstantinou, J.; Fry, C.S.; Watowich, S.J. Small Molecule Nicotinamide N-Methyltransferase Inhibitor Activates Senescent Muscle Stem Cells and Improves Regenerative Capacity of Aged Skeletal Muscle. Biochem. Pharmacol. 2019, 163, 481–492. [Google Scholar] [CrossRef]
- Altin, K.; Tabassum, C.; Uddin Mohammed, S.; Junaid Rashad, R.; Ramsden David, B.; Geshanthi, H.; Fábio, K.; Linda, P. Parsons Richard B High Expression of Nicotinamide N-Methyltransferase in Patients with Sporadic Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 1769–1781. [Google Scholar]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT Promotes Epigenetic Remodeling in Cancer by Creating a Metabolic Methylation Sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef]
- Ramsden, D.B.; Waring, R.H.; Barlow, D.J.; Parsons, R.B. Nicotinamide N-Methyltransferase in Health and Cancer. Int. J. Tryptophan Res. 2017, 10, 117864691769173. [Google Scholar] [CrossRef]
- Lu, X.M.; Long, H. Nicotinamide N-Methyltransferase as a Potential Marker for Cancer. NEO 2018, 65, 656–663. [Google Scholar] [CrossRef]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.; et al. Nicotinamide N-Methyltransferase Knockdown Protects against Diet-Induced Obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef]
- Guan, X.-X.; Zhu, X.-J.; Deng, Z.-H.; Zeng, Y.-R.; Liu, J.-R.; Li, J.-H. The Association between Nicotinamide N-Methyltransferase Gene Polymorphisms and Primary Hypertension in Chinese Han Population. AIMS Bioeng. 2021, 8, 130–139. [Google Scholar] [CrossRef]
- Zhu, X.-J.; Lin, Y.-J.; Chen, W.; Wang, Y.-H.; Qiu, L.-Q.; Cai, C.-X.; Xiong, Q.; Chen, F.; Chen, L.-H.; Zhou, Q.; et al. Physiological Study on Association between Nicotinamide N-Methyltransferase Gene Polymorphisms and Hyperlipidemia. BioMed Res. Int. 2016, 2016, 7521942. [Google Scholar] [CrossRef]
- Souto, J.C.; Blanco-Vaca, F.; Soria, J.M.; Buil, A.; Almasy, L.; Ordoñez-Llanos, J.; Mª Martín-Campos, J.; Lathrop, M.; Stone, W.; Blangero, J.; et al. A Genomewide Exploration Suggests a New Candidate Gene at Chromosome 11q23 as the Major Determinant of Plasma Homocysteine Levels: Results from the GAIT Project. Am. J. Hum. Genet. 2005, 76, 925–933. [Google Scholar] [CrossRef]
- Dinavahi, R.; Falkner, B. Relationship of Homocysteine with Cardiovascular Disease and Blood Pressure. J. Clin. Hypertens 2004, 6, 494–498; quiz 499–500. [Google Scholar] [CrossRef]
- Petras, M.; Tatarkova, Z.; Kovalska, M.; Mokra, D.; Dobrota, D.; Lehotsky, J.; Drgova, A. Hyperhomocysteinemia as a Risk Factor for the Neuronal System Disorders. J. Physiol. Pharmacol. 2014, 65, 15–23. [Google Scholar]
- Li, S.; Wei, J.; Zhang, C.; Li, X.; Meng, W.; Mo, X.; Zhang, Q.; Liu, Q.; Ren, K.; Du, R.; et al. Cell-Derived Microparticles in Patients with Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis. Cell Physiol. Biochem. 2016, 39, 2439–2450. [Google Scholar] [CrossRef]
- Hasan, T.; Arora, R.; Bansal, A.K.; Bhattacharya, R.; Sharma, G.S.; Singh, L.R. Disturbed Homocysteine Metabolism Is Associated with Cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Kaplan, P.; Tatarkova, Z.; Sivonova, M.K.; Racay, P.; Lehotsky, J. Homocysteine and Mitochondria in Cardiovascular and Cerebrovascular Systems. Int. J. Mol. Sci. 2020, 21, 7698. [Google Scholar] [CrossRef]
- Familtseva, A.; Chaturvedi, P.; Kalani, A.; Jeremic, N.; Metreveli, N.; Kunkel, G.H.; Tyagi, S.C. Toll-like Receptor 4 Mutation Suppresses Hyperhomocysteinemia-Induced Hypertension. Am. J. Physiol. Cell Physiol. 2016, 311, C596–C606. [Google Scholar] [CrossRef]
- Ansari, R.; Mahta, A.; Mallack, E.; Luo, J.J. Hyperhomocysteinemia and Neurologic Disorders: A Review. J. Clin. Neurol. 2014, 10, 281–288. [Google Scholar] [CrossRef]
- Hu, X.-W.; Qin, S.-M.; Li, D.; Hu, L.-F.; Liu, C.-F. Elevated Homocysteine Levels in Levodopa-Treated Idiopathic Parkinson’s Disease: A Meta-Analysis. Acta Neurol. Scand. 2013, 128, 73–82. [Google Scholar] [CrossRef]
- Price, B.R.; Wilcock, D.M.; Weekman, E.M. Hyperhomocysteinemia as a Risk Factor for Vascular Contributions to Cognitive Impairment and Dementia. Front. Aging Neurosci. 2018, 10, 350. [Google Scholar] [CrossRef]
- Bhargava, S.; Bhandari, A.; Choudhury, S. Role of Homocysteine in Cognitive Impairement and Alzheimer’s Disease. Indian. J. Clin. Biochem. 2018, 33, 16–20. [Google Scholar] [CrossRef]
- Ala, O.A.; Akintunde, A.A.; Ikem, R.T.; Kolawole, B.A.; Ala, O.O.; Adedeji, T.A. Association between Insulin Resistance and Total Plasma Homocysteine Levels in Type 2 Diabetes Mellitus Patients in South West Nigeria. Diabetes Metab. Syndr. 2017, 11 (Suppl. 2), S803–S809. [Google Scholar] [CrossRef]
- Jakubowski, H. Protein N-Homocysteinylation and Colorectal Cancer. Trends Cancer 2019, 5, 7–10. [Google Scholar] [CrossRef]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef]
- Goody, M.F.; Henry, C.A. A Need for NAD+ in Muscle Development, Homeostasis, and Aging. Skelet. Muscle 2018, 8, 9. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ Metabolism and Its Roles in Cellular Processes during Ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Frederick, D.W.; Loro, E.; Liu, L.; Davila, A.; Chellappa, K.; Silverman, I.M.; Quinn, W.J.; Gosai, S.J.; Tichy, E.D.; Davis, J.G.; et al. Loss of NAD Homeostasis Leads to Progressive and Reversible Degeneration of Skeletal Muscle. Cell Metab. 2016, 24, 269–282. [Google Scholar] [CrossRef]
- Mori, V.; Amici, A.; Mazzola, F.; Di Stefano, M.; Conforti, L.; Magni, G.; Ruggieri, S.; Raffaelli, N.; Orsomando, G. Metabolic Profiling of Alternative NAD Biosynthetic Routes in Mouse Tissues. PLoS ONE 2014, 9, e113939. [Google Scholar] [CrossRef]
- Berger, F.; Lau, C.; Dahlmann, M.; Ziegler, M. Subcellular Compartmentation and Differential Catalytic Properties of the Three Human Nicotinamide Mononucleotide Adenylyltransferase Isoforms. J. Biol. Chem. 2005, 280, 36334–36341. [Google Scholar] [CrossRef]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ Metabolism: Pathophysiologic Mechanisms and Therapeutic Potential. Sig. Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 Inhibition Increases Mitochondrial Metabolism through SIRT1 Activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef]
- Barbosa, M.T.P.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The Enzyme CD38 (a NAD Glycohydrolase, EC 3.2.2.5) Is Necessary for the Development of Diet-Induced Obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef]
- Li, F.; Chong, Z.; Maiese, K. Cell Life Versus Cell Longevity: The Mysteries Surrounding the NAD+ Precursor Nicotinamide. CMC 2006, 13, 883–895. [Google Scholar] [CrossRef]
- Liu, J.-R.; Deng, Z.-H.; Zhu, X.-J.; Zeng, Y.-R.; Guan, X.-X.; Li, J.-H. Roles of Nicotinamide N-Methyltransferase in Obesity and Type 2 Diabetes. Biomed. Res. Int. 2021, 2021, 9924314. [Google Scholar] [CrossRef]
- Plazar, N.; Jurdana, M. Hyperhomocysteinemia and the Role of B Vitamins in Cancer. Radiol. Oncol. 2010, 44, 79–85. [Google Scholar] [CrossRef]
- Van Guldener, C.; Nanayakkara, P.W.B.; Stehouwer, C.D.A. Homocysteine and Blood Pressure. Curr. Sci. Inc. 2003, 5, 26–31. [Google Scholar] [CrossRef]
- Zhang, J.; Shu, Y.; Qu, Y.; Zhang, L.; Chu, T.; Zheng, Y.; Zhao, H. C-Myb Plays an Essential Role in the Protective Function of IGF-1 on Cytotoxicity Induced by Aβ25-35 via the PI3K/Akt Pathway. J. Mol. Neurosci. 2017, 63, 412–418. [Google Scholar] [CrossRef]
- Elsherbiny, N.M.; Sharma, I.; Kira, D.; Alhusban, S.; Samra, Y.A.; Jadeja, R.; Martin, P.; Al-Shabrawey, M.; Tawfik, A. Homocysteine Induces Inflammation in Retina and Brain. Biomolecules 2020, 10, 393. [Google Scholar] [CrossRef]
- Koklesova, L.; Samec, M.; Liskova, A.; Zhai, K.; Büsselberg, D.; Giordano, F.A.; Kubatka, P.; Golunitschaja, O. Mitochondrial Impairments in Aetiopathology of Multifactorial Diseases: Common Origin but Individual Outcomes in Context of 3P Medicine. EPMA J. 2021, 12, 27–40. [Google Scholar] [CrossRef]
- Koklesova, L.; Liskova, A.; Samec, M.; Zhai, K.; AL-Ishaq, R.K.; Bugos, O.; Šudomová, M.; Biringer, K.; Pec, M.; Adamkov, M.; et al. Protective Effects of Flavonoids against Mitochondriopathies and Associated Pathologies: Focus on the Predictive Approach and Personalized Prevention. Int. J. Mol. Sci. 2021, 22, 8649. [Google Scholar] [CrossRef]
- McCully, K.S. Review: Chemical Pathology of Homocysteine VI. Aging, Cellular Senescence, and Mitochondrial Dysfunction. Ann. Clin. Lab. Sci. 2018, 48, 677–687. [Google Scholar]
- Wang, L.; Niu, H.; Zhang, J. Homocysteine Induces Mitochondrial Dysfunction and Oxidative Stress in Myocardial Ischemia/Reperfusion Injury through Stimulating ROS Production and the ERK1/2 Signaling Pathway. Exp. Ther. Med. 2020, 20, 938–944. [Google Scholar] [CrossRef]
- Veeranki, S.; Winchester, L.J.; Tyagi, S.C. Hyperhomocysteinemia Associated Skeletal Muscle Weakness Involves Mitochondrial Dysfunction and Epigenetic Modifications. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2015, 1852, 732–741. [Google Scholar] [CrossRef]
- Timkova, V.; Tatarkova, Z.; Lehotsky, J.; Racay, P.; Dobrota, D.; Kaplan, P. Effects of Mild Hyperhomocysteinemia on Electron Transport Chain Complexes, Oxidative Stress, and Protein Expression in Rat Cardiac Mitochondria. Mol. Cell Biochem. 2016, 411, 261–270. [Google Scholar] [CrossRef]
- Zhang, Z.; Wei, C.; Zhou, Y.; Yan, T.; Wang, Z.; Li, W.; Zhao, L. Homocysteine Induces Apoptosis of Human Umbilical Vein Endothelial Cells via Mitochondrial Dysfunction and Endoplasmic Reticulum Stress. Oxidative Med. Cell. Longev. 2017, 2017, 5736506. [Google Scholar] [CrossRef]
- Bhattacharjee, N.; Borah, A. Oxidative Stress and Mitochondrial Dysfunction Are the Underlying Events of Dopaminergic Neurodegeneration in Homocysteine Rat Model of Parkinson’s Disease. Neurochem. Int. 2016, 101, 48–55. [Google Scholar] [CrossRef]
- Mohamad Kamal, N.S.; Safuan, S.; Shamsuddin, S.; Foroozandeh, P. Aging of the Cells: Insight into Cellular Senescence and Detection Methods. Eur. J. Cell Biol. 2020, 99, 151108. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Yousefzadeh, M.J.; Niedernhofer, L.J.; Robbins, P.D.; Zhu, Y. Cellular Senescence: A Key Therapeutic Target in Aging and Diseases. J. Clin. Investig. 2022, 132, e158450. [Google Scholar] [CrossRef]
- Bulbiankova, D.; Díaz-Puertas, R.; Álvarez-Martínez, F.J.; Herranz-López, M.; Barrajón-Catalán, E.; Micol, V. Hallmarks and Biomarkers of Skin Senescence: An Updated Review of Skin Senotherapeutics. Antioxidants 2023, 12, 444. [Google Scholar] [CrossRef]
- Cho, J.H.; Kim, E.-C.; Son, Y.; Lee, D.-W.; Park, Y.S.; Choi, J.H.; Cho, K.-H.; Kwon, K.-S.; Kim, J.-R. CD9 Induces Cellular Senescence and Aggravates Atherosclerotic Plaque Formation. Cell Death Differ. 2020, 27, 2681–2696. [Google Scholar] [CrossRef]
- Saez-Atienzar, S.; Masliah, E. Cellular Senescence and Alzheimer Disease: The Egg and the Chicken Scenario. Nat. Rev. Neurosci. 2020, 21, 433–444. [Google Scholar] [CrossRef]
- Chen, S.; Wu, P.; Zhou, L.; Shen, Y.; Li, Y.; Song, H. Relationship between Increase of Serum Homocysteine Caused by Smoking and Oxidative Damage in Elderly Patients with Cardiovascular Disease. Int. J. Clin. Exp. Med. 2015, 8, 4446–4454. [Google Scholar]
- Fagagna, F.; D’adda, D.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA Damage Checkpoint Response in Telomere-Initiated Senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Zhang, D.; Sun, X.; Liu, J.; Xie, X.; Cui, W.; Zhu, Y. Homocysteine Accelerates Senescence of Endothelial Cells via DNA Hypomethylation of Human Telomerase Reverse Transcriptase. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 71–78. [Google Scholar] [CrossRef]
- Hossain, G.S.; Van Thienen, J.V.; Werstuck, G.H.; Zhou, J.; Sood, S.K.; Dickhout, J.G.; De Koning, A.B.L.; Tang, D.; Wu, D.; Falk, E.; et al. TDAG51 Is Induced by Homocysteine, Promotes Detachment-Mediated Programmed Cell Death, and Contributes to the Development of Atherosclerosis in Hyperhomocysteinemia. J. Biol. Chem. 2003, 278, 30317–30327. [Google Scholar] [CrossRef]
- Zhou, J.; Werstuck, G.H.; Lhoták, S.; Shi, Y.Y.; Tedesco, V.; Trigatti, B.; Dickhout, J.; Majors, A.K.; DiBello, P.M.; Jacobsen, D.W.; et al. Hyperhomocysteinemia Induced by Methionine Supplementation Does Not Independently Cause Atherosclerosis in C57BL/6J Mice. FASEB J. 2008, 22, 2569–2578. [Google Scholar] [CrossRef]
- Wang, H.; Yoshizumi, M.; Lai, K.; Tsai, J.C.; Perrella, M.A.; Haber, E.; Lee, M.E. Inhibition of Growth and P21ras Methylation in Vascular Endothelial Cells by Homocysteine but Not Cysteine. J. Biol. Chem. 1997, 272, 25380–25385. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, X.; Yang, F.; Chapman, G.B.; Durante, W.; Sibinga, N.E.S.; Schafer, A.I. Cyclin A Transcriptional Suppression Is the Major Mechanism Mediating Homocysteine-Induced Endothelial Cell Growth Inhibition. Blood 2002, 99, 939–945. [Google Scholar] [CrossRef]
- Kałużna-Czaplińska, J.; Żurawicz, E.; Michalska, M.; Rynkowski, J. A Focus on Homocysteine in Autism. Acta Biochim. Pol. 2013, 60, 137–142. [Google Scholar] [CrossRef]
- Mattson, M.P.; Shea, T.B. Folate and Homocysteine Metabolism in Neural Plasticity and Neurodegenerative Disorders. Trends Neurosci. 2003, 26, 137–146. [Google Scholar] [CrossRef]
- Ho, P.I.; Ortiz, D.; Rogers, E.; Shea, T.B. Multiple Aspects of Homocysteine Neurotoxicity: Glutamate Excitotoxicity, Kinase Hyperactivation and DNA Damage. J. Neurosci. Res. 2002, 70, 694–702. [Google Scholar] [CrossRef]
- Csekes, E.; Račková, L. Skin Aging, Cellular Senescence and Natural Polyphenols. Int. J. Mol. Sci. 2021, 22, 12641. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: A Concept in Redox Biology and Medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The Role of Antioxidants in the Chemistry of Oxidative Stress: A Review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Papaconstantinou, J. The Role of Signaling Pathways of Inflammation and Oxidative Stress in Development of Senescence and Aging Phenotypes in Cardiovascular Disease. Cells 2019, 8, 1383. [Google Scholar] [CrossRef]
- Kao, Y.-C.; Ho, P.-C.; Tu, Y.-K.; Jou, I.-M.; Tsai, K.-J. Lipids and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1505. [Google Scholar] [CrossRef]
- Sibrian-Vazquez, M.; Escobedo, J.O.; Lim, S.; Samoei, G.K.; Strongin, R.M. Homocystamides Promote Free-Radical and Oxidative Damage to Proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 551–554. [Google Scholar] [CrossRef]
- Lehotský, J.; Tothová, B.; Kovalská, M.; Dobrota, D.; Beňová, A.; Kalenská, D.; Kaplán, P. Role of Homocysteine in the Ischemic Stroke and Development of Ischemic Tolerance. Front. Neurosci. 2016, 10, 538. [Google Scholar] [CrossRef]
- Çelik, N.; Vurmaz, A.; Kahraman, A. Protective Effect of Quercetin on Homocysteine-Induced Oxidative Stress. Nutrition 2017, 33, 291–296. [Google Scholar] [CrossRef]
- Papatheodorou, L.; Weiss, N. Vascular Oxidant Stress and Inflammation in Hyperhomocysteinemia. Antioxid. Redox Signal. 2007, 9, 1941–1958. [Google Scholar] [CrossRef]
- Bharathi Devi, S.R.; Suganeswari, G.; Sharma, T.; Thennarasu, M.; Angayarkanni, N. Homocysteine Induces Oxidative Stress in Young Adult Central Retinal Vein Occlusion. Br. J. Ophthalmol. 2012, 96, 1122–1126. [Google Scholar] [CrossRef]
- Rehman, T.; Shabbir, M.A.; Inam-Ur-Raheem, M.; Manzoor, M.F.; Ahmad, N.; Liu, Z.-W.; Ahmad, M.H.; Siddeeg, A.; Abid, M.; Aadil, R.M. Cysteine and Homocysteine as Biomarker of Various Diseases. Food Sci. Nutr. 2020, 8, 4696–4707. [Google Scholar] [CrossRef]
- Tawfik, A.; Samra, Y.A.; Elsherbiny, N.M.; Al-Shabrawey, M. Implication of Hyperhomocysteinemia in Blood Retinal Barrier (BRB) Dysfunction. Biomolecules 2020, 10, 1119. [Google Scholar] [CrossRef]
- Aghayan, S.S.; Farajzadeh, A.; Bagheri-Hosseinabadi, Z.; Fadaei, H.; Yarmohammadi, M.; Jafarisani, M. Elevated Homocysteine, as a Biomarker of Cardiac Injury, in Panic Disorder Patients Due to Oxidative Stress. Brain Behav 2020, 10, e01851. [Google Scholar] [CrossRef]
- Scherer, E.B.S.; Loureiro, S.O.; Vuaden, F.C.; da Cunha, A.A.; Schmitz, F.; Kolling, J.; Savio, L.E.B.; Bogo, M.R.; Bonan, C.D.; Netto, C.A.; et al. Mild Hyperhomocysteinemia Increases Brain Acetylcholinesterase and Proinflammatory Cytokine Levels in Different Tissues. Mol. Neurobiol. 2014, 50, 589–596. [Google Scholar] [CrossRef]
- Yang, X.; Gao, F.; Liu, Y. Association of Homocysteine with Immunological-Inflammatory and Metabolic Laboratory Markers and Factors in Relation to Hyperhomocysteinaemia in Rheumatoid Arthritis. Clin. Exp. Rheumatol. 2015, 33, 900–903. [Google Scholar]
- Lipton, S.A.; Kim, W.K.; Choi, Y.B.; Kumar, S.; D’Emilia, D.M.; Rayudu, P.V.; Arnelle, D.R.; Stamler, J.S. Neurotoxicity Associated with Dual Actions of Homocysteine at the N-Methyl-D-Aspartate Receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928. [Google Scholar] [CrossRef]
- Moshal, K.S.; Metreveli, N.; Frank, I.; Tyagi, S.C. Mitochondrial MMP Activation, Dysfunction and Arrhythmogenesis in Hyperhomocysteinemia. Curr. Vasc. Pharm. 2008, 6, 84–92. [Google Scholar] [CrossRef]
- Steed, M.M.; Tyagi, S.C. Mechanisms of Cardiovascular Remodeling in Hyperhomocysteinemia. Antioxid. Redox Signal. 2011, 15, 1927–1943. [Google Scholar] [CrossRef]
- Vacek, T.P.; Vacek, J.C.; Tyagi, S.C. Mitochondrial Mitophagic Mechanisms of Myocardial Matrix Metabolism and Remodelling. Arch. Physiol. Biochem. 2012, 118, 31–42. [Google Scholar] [CrossRef]
- Wu, J.; Jin, Z.; Zheng, H.; Yan, L.-J. Sources and Implications of NADH/NAD(+) Redox Imbalance in Diabetes and Its Complications. Diabetes Metab. Syndr. Obes. 2016, 9, 145–153. [Google Scholar] [CrossRef]
- Lin, Q.; Zuo, W.; Liu, Y.; Wu, K.; Liu, Q. NAD+ and Cardiovascular Diseases. Clin. Chim. Acta 2021, 515, 104–110. [Google Scholar] [CrossRef]
- Schöndorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef]
- Bordone, L.; Motta, M.C.; Picard, F.; Robinson, A.; Jhala, U.S.; Apfeld, J.; McDonagh, T.; Lemieux, M.; McBurney, M.; Szilvasi, A.; et al. Sirt1 Regulates Insulin Secretion by Repressing UCP2 in Pancreatic Beta Cells. PLoS Biol. 2006, 4, e31. [Google Scholar] [CrossRef]
- Pfluger, P.T.; Herranz, D.; Velasco-Miguel, S.; Serrano, M.; Tschöp, M.H. Sirt1 Protects against High-Fat Diet-Induced Metabolic Damage. Proc. Natl. Acad. Sci. USA 2008, 105, 9793–9798. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Gerdts, J.; Brace, E.J.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. SARM1 Activation Triggers Axon Degeneration Locally via NAD+ Destruction. Science 2015, 348, 453–457. [Google Scholar] [CrossRef]
- Kauppinen, T.M.; Suh, S.W.; Higashi, Y.; Berman, A.E.; Escartin, C.; Won, S.J.; Wang, C.; Cho, S.-H.; Gan, L.; Swanson, R.A. Poly(ADP-Ribose)Polymerase-1 Modulates Microglial Responses to Amyloid β. J. Neuroinflammation 2011, 8, 152. [Google Scholar] [CrossRef]
- Mandir, A.S.; Przedborski, S.; Jackson-Lewis, V.; Wang, Z.Q.; Simbulan-Rosenthal, C.M.; Smulson, M.E.; Hoffman, B.E.; Guastella, D.B.; Dawson, V.L.; Dawson, T.M. Poly(ADP-Ribose) Polymerase Activation Mediates 1-Methyl-4-Phenyl-1, 2,3,6-Tetrahydropyridine (MPTP)-Induced Parkinsonism. Proc. Natl. Acad. Sci. USA 1999, 96, 5774–5779. [Google Scholar] [CrossRef]
- Zhou, C.; Yang, X.; Hua, X.; Liu, J.; Fan, M.; Li, G.; Song, J.; Xu, T.; Li, Z.; Guan, Y.; et al. Hepatic NAD+ Deficiency as a Therapeutic Target for Non-alcoholic Fatty Liver Disease in Ageing. Br. J. Pharmacol. 2016, 173, 2352–2368. [Google Scholar] [CrossRef]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G.J. Age-Associated Changes in Oxidative Stress and NAD+ Metabolism In Human Tissue. PLoS ONE 2012, 7, e42357. [Google Scholar] [CrossRef]
- Zhu, X.-H.; Lu, M.; Lee, B.-Y.; Ugurbil, K.; Chen, W. In Vivo NAD Assay Reveals the Intracellular NAD Contents and Redox State in Healthy Human Brain and Their Age Dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef]
- Clement, J.; Wong, M.; Poljak, A.; Sachdev, P.; Braidy, N. The Plasma NAD+ Metabolome Is Dysregulated in “Normal” Aging. Rejuvenation Res. 2019, 22, 121–130. [Google Scholar] [CrossRef]
- Lamb, D.A.; Moore, J.H.; Mesquita, P.H.C.; Smith, M.A.; Vann, C.G.; Osburn, S.C.; Fox, C.D.; Lopez, H.L.; Ziegenfuss, T.N.; Huggins, K.W.; et al. Resistance Training Increases Muscle NAD+ and NADH Concentrations as Well as NAMPT Protein Levels and Global Sirtuin Activity in Middle-Aged, Overweight, Untrained Individuals. Aging 2020, 12, 9447–9460. [Google Scholar] [CrossRef]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de Novo NAD+ Synthesis Specifies Immune Function in Aging and Inflammation. Nat. Immunol. 2019, 20, 50–63. [Google Scholar] [CrossRef]
- Yu, P.; Cai, X.; Liang, Y.; Wang, M.; Yang, W. Roles of NAD+ and Its Metabolites Regulated Calcium Channels in Cancer. Molecules 2020, 25, 4826. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD+ in Aging: Molecular Mechanisms and Translational Implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef]
- Xie, X.; Yu, H.; Wang, Y.; Zhou, Y.; Li, G.; Ruan, Z.; Li, F.; Wang, X.; Liu, H.; Zhang, J. Nicotinamide N-Methyltransferase Enhances the Capacity of Tumorigenesis Associated with the Promotion of Cell Cycle Progression in Human Colorectal Cancer Cells. Arch. Biochem. Biophys. 2014, 564, 52–66. [Google Scholar] [CrossRef]
- Eckert, M.A.; Coscia, F.; Chryplewicz, A.; Chang, J.W.; Hernandez, K.M.; Pan, S.; Tienda, S.M.; Nahotko, D.A.; Li, G.; Blaženović, I.; et al. Proteomics Reveals NNMT as a Master Metabolic Regulator of Cancer-Associated Fibroblasts. Nature 2019, 569, 723–728. [Google Scholar] [CrossRef]
- Takahashi, R.; Kanda, T.; Komatsu, M.; Itoh, T.; Minakuchi, H.; Urai, H.; Kuroita, T.; Shigaki, S.; Tsukamoto, T.; Higuchi, N.; et al. The Significance of NAD + Metabolites and Nicotinamide N-Methyltransferase in Chronic Kidney Disease. Sci. Rep. 2022, 12, 6398. [Google Scholar] [CrossRef]
- Sun, W.; Zou, Y.; Cai, Z.; Huang, J.; Hong, X.; Liang, Q.; Jin, W. Overexpression of NNMT in Glioma Aggravates Tumor Cell Progression: An Emerging Therapeutic Target. Cancers 2022, 14, 3538. [Google Scholar] [CrossRef]
- Cui, Y.; Yang, D.; Wang, W.; Zhang, L.; Liu, H.; Ma, S.; Guo, W.; Yao, M.; Zhang, K.; Li, W.; et al. Nicotinamide N-Methyltransferase Decreases 5-Fluorouracil Sensitivity in Human Esophageal Squamous Cell Carcinoma through Metabolic Reprogramming and Promoting the Warburg Effect. Mol. Carcinog. 2020, 59, 940–954. [Google Scholar] [CrossRef]
- Cantoni, G.L. Methylation of Nicotinamide with Soluble Enzyme System from Rat Liver. J. Biol. Chem. 1951, 189, 203–216. [Google Scholar] [CrossRef]
- Marton, O.; Koltai, E.; Nyakas, C.; Bakonyi, T.; Zenteno-Savin, T.; Kumagai, S.; Goto, S.; Radak, Z. Aging and Exercise Affect the Level of Protein Acetylation and SIRT1 Activity in Cerebellum of Male Rats. Biogerontology 2010, 11, 679–686. [Google Scholar] [CrossRef]
- Haigis, M.C.; Guarente, L.P. Mammalian Sirtuins—Emerging Roles in Physiology, Aging, and Calorie Restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Vasudevan, P.; Zhong, L.; Kim, G.; Samant, S.; Parekh, V.; Pillai, V.B.; Ravindra, P.V.; Gupta, M.; Jeevanandam, V.; et al. The Sirtuin SIRT6 Blocks IGF-Akt Signaling and Development of Cardiac Hypertrophy by Targeting c-Jun. Nat. Med. 2012, 18, 1643–1650. [Google Scholar] [CrossRef]
- Trammell, S.A.J.; Brenner, C. NNMT: A Bad Actor in Fat Makes Good in Liver. Cell Metab. 2015, 22, 200–201. [Google Scholar] [CrossRef]
- Kannt, A.; Pfenninger, A.; Teichert, L.; Tönjes, A.; Dietrich, A.; Schön, M.R.; Klöting, N.; Blüher, M. Association of Nicotinamide-N-Methyltransferase mRNA Expression in Human Adipose Tissue and the Plasma Concentration of Its Product, 1-Methylnicotinamide, with Insulin Resistance. Diabetologia 2015, 58, 799–808. [Google Scholar] [CrossRef]
- Haigis, M.C.; Sinclair, D.A. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef]
- Guarente, L. Calorie Restriction and Sirtuins Revisited. Genes. Dev. 2013, 27, 2072–2085. [Google Scholar] [CrossRef]
- Satoh, A.; Stein, L.; Imai, S. The Role of Mammalian Sirtuins in the Regulation of Metabolism, Aging, and Longevity. Handb. Exp. Pharmacol. 2011, 206, 125–162. [Google Scholar] [CrossRef]
- Das, A.; Huang, G.X.; Bonkowski, M.S.; Longchamp, A.; Li, C.; Schultz, M.B.; Kim, L.-J.; Osborne, B.; Joshi, S.; Lu, Y.; et al. Impairment of an Endothelial NAD+-H2S Signaling Network Is a Reversible Cause of Vascular Aging. Cell 2018, 173, 74–89.e20. [Google Scholar] [CrossRef]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide Mononucleotide, a Key NAD+ Intermediate, Treats the Pathophysiology of Diet- and Age-Induced Diabetes in Mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef]
- Pajk, M.; Cselko, A.; Varga, C.; Posa, A.; Tokodi, M.; Boldogh, I.; Goto, S.; Radak, Z. Exogenous Nicotinamide Supplementation and Moderate Physical Exercise Can Attenuate the Aging Process in Skeletal Muscle of Rats. Biogerontology 2017, 18, 593–600. [Google Scholar] [CrossRef]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De Novo NAD+ Synthesis Enhances Mitochondrial Function and Improves Health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef]
- Rogina, B.; Helfand, S.L. Sir2 Mediates Longevity in the Fly through a Pathway Related to Calorie Restriction. Proc. Natl. Acad. Sci. USA 2004, 101, 15998–16003. [Google Scholar] [CrossRef]
- Chopra, V.; Quinti, L.; Kim, J.; Vollor, L.; Narayanan, K.L.; Edgerly, C.; Cipicchio, P.M.; Lauver, M.A.; Choi, S.H.; Silverman, R.B.; et al. The Sirtuin 2 Inhibitor AK-7 Is Neuroprotective in Huntington’s Disease Mouse Models. Cell Rep. 2012, 2, 1492–1497. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Tarragó, M.G.; Chini, E.N. NAD and the Aging Process: Role in Life, Death and Everything in between. Mol. Cell. Endocrinol. 2017, 455, 62–74. [Google Scholar] [CrossRef]
- Estep, P.W.; Warner, J.B.; Bulyk, M.L. Short-Term Calorie Restriction in Male Mice Feminizes Gene Expression and Alters Key Regulators of Conserved Aging Regulatory Pathways. PLoS ONE 2009, 4, e5242. [Google Scholar] [CrossRef]
- Ye, X.; Li, M.; Hou, T.; Gao, T.; Zhu, W.-G.; Yang, Y. Sirtuins in Glucose and Lipid Metabolism. Oncotarget 2017, 8, 1845–1859. [Google Scholar] [CrossRef]
- Hong, S.; Moreno-Navarrete, J.M.; Wei, X.; Kikukawa, Y.; Tzameli, I.; Prasad, D.; Lee, Y.; Asara, J.M.; Fernandez-Real, J.M.; Maratos-Flier, E.; et al. Nicotinamide N-Methyltransferase Regulates Hepatic Nutrient Metabolism through Sirt1 Protein Stabilization. Nat. Med. 2015, 21, 887–894. [Google Scholar] [CrossRef]
- Liu, K.Y.; Mistry, R.J.; Aguirre, C.A.; Fasouli, E.S.; Thomas, M.G.; Klamt, F.; Ramsden, D.B.; Parsons, R.B. Nicotinamide N-Methyltransferase Increases Complex I Activity in SH-SY5Y Cells via Sirtuin 3. Biochem. Biophys. Res. Commun. 2015, 467, 491–496. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, J.; Wu, W.; Xie, S.; Yu, H.; Li, G.; Zhu, T.; Li, F.; Lu, J.; Wang, G.Y.; et al. Nicotinamide N-Methyltransferase Enhances Chemoresistance in Breast Cancer through SIRT1 Protein Stabilization. Breast Cancer Res. 2019, 21, 64. [Google Scholar] [CrossRef]
- You, Z.; Liu, Y.; Liu, X. Nicotinamide N-methyltransferase Enhances the Progression of Prostate Cancer by Stabilizing Sirtuin 1. Oncol. Lett. 2018, 15, 9195–9201. [Google Scholar] [CrossRef]
- Schmeisser, K.; Mansfeld, J.; Kuhlow, D.; Weimer, S.; Priebe, S.; Heiland, I.; Birringer, M.; Groth, M.; Segref, A.; Kanfi, Y.; et al. Role of Sirtuins in Lifespan Regulation Is Linked to Methylation of Nicotinamide. Nat. Chem. Biol. 2013, 9, 693–700. [Google Scholar] [CrossRef]
- Parsons, R.B.; Aravindan, S.; Kadampeswaran, A.; Evans, E.A.; Sandhu, K.K.; Levy, E.R.; Thomas, M.G.; Austen, B.M.; Ramsden, D.B. The Expression of Nicotinamide N-Methyltransferase Increases ATP Synthesis and Protects SH-SY5Y Neuroblastoma Cells against the Toxicity of Complex I Inhibitors. Biochem. J. 2011, 436, 145–155. [Google Scholar] [CrossRef]
- Larsen, S.C.; Hendriks, I.A.; Lyon, D.; Jensen, L.J.; Nielsen, M.L. Systems-Wide Analysis of Serine ADP-Ribosylation Reveals Widespread Occurrence and Site-Specific Overlap with Phosphorylation. Cell Rep. 2018, 24, 2493–2505.e4. [Google Scholar] [CrossRef]
- De Murcia, G.; De Murcia, J.M. Poly(ADP-Ribose) Polymerase: A Molecular Nick-Sensor. Trends Biochem. Sci. 1994, 19, 172–176. [Google Scholar] [CrossRef]
- Summers, D.W.; Gibson, D.A.; DiAntonio, A.; Milbrandt, J. SARM1-Specific Motifs in the TIR Domain Enable NAD+ Loss and Regulate Injury-Induced SARM1 Activation. Proc. Natl. Acad. Sci. USA 2016, 113, E6271–E6280. [Google Scholar] [CrossRef]
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD + Cleavage Activity That Promotes Pathological Axonal Degeneration. Neuron 2017, 93, 1334–1343.e5. [Google Scholar] [CrossRef]
- Saldeen, J.; Welsh, N. Nicotinamide-Induced Apoptosis in Insulin Producing Cells Is Associated with Cleavage of Poly(ADP-Ribose) Polymerase. Mol. Cell Endocrinol. 1998, 139, 99–107. [Google Scholar] [CrossRef]
- Slominska, E.M.; Kowalik, K.; Smolenski, R.T.; Szolkiewicz, M.; Rutkowski, P.; Rutkowski, B.; Swierczynski, J. Accumulation of Poly(ADP-Ribose) Polymerase Inhibitors in Children with Chronic Renal Failure. Pediatr. Nephrol. 2006, 21, 800–806. [Google Scholar] [CrossRef]
- Zhang, J. Are Poly(ADP-ribosyl)Ation by PARP-1 and Deacetylation by Sir2 Linked? BioEssays 2003, 25, 808–814. [Google Scholar] [CrossRef]
- Pirinen, E.; Cantó, C.; Jo, Y.S.; Morato, L.; Zhang, H.; Menzies, K.J.; Williams, E.G.; Mouchiroud, L.; Moullan, N.; Hagberg, C.; et al. Pharmacological Inhibition of Poly(ADP-Ribose) Polymerases Improves Fitness and Mitochondrial Function in Skeletal Muscle. Cell Metab. 2014, 19, 1034–1041. [Google Scholar] [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) Induces a Pseudohypoxic State Disrupting Nuclear-Mitochondrial Communication during Aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A High-Fat Diet and NAD(+) Activate Sirt1 to Rescue Premature Aging in Cockayne Syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.-S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD+/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef]
- Wu, M.-F.; Yin, J.-H.; Hwang, C.-S.; Tang, C.-M.; Yang, D.-I. NAD Attenuates Oxidative DNA Damages Induced by Amyloid Beta-Peptide in Primary Rat Cortical Neurons. Free Radic. Res. 2014, 48, 794–805. [Google Scholar] [CrossRef]
- Gariani, K.; Ryu, D.; Menzies, K.J.; Yi, H.-S.; Stein, S.; Zhang, H.; Perino, A.; Lemos, V.; Katsyuba, E.; Jha, P.; et al. Inhibiting Poly ADP-Ribosylation Increases Fatty Acid Oxidation and Protects against Fatty Liver Disease. J. Hepatol. 2017, 66, 132–141. [Google Scholar] [CrossRef]
- D’Andrea, F.P.; Safwat, A.; Kassem, M.; Gautier, L.; Overgaard, J.; Horsman, M.R. Cancer Stem Cell Overexpression of Nicotinamide N-Methyltransferase Enhances Cellular Radiation Resistance. Radiother. Oncol. 2011, 99, 373–378. [Google Scholar] [CrossRef]
- Lenglet, A.; Liabeuf, S.; Bodeau, S.; Louvet, L.; Mary, A.; Boullier, A.; Lemaire-Hurtel, A.; Jonet, A.; Sonnet, P.; Kamel, S.; et al. N-Methyl-2-Pyridone-5-Carboxamide (2PY)—Major Metabolite of Nicotinamide: An Update on an Old Uremic Toxin. Toxins 2016, 8, 339. [Google Scholar] [CrossRef]
- Chini, C.; Hogan, K.A.; Warner, G.M.; Tarragó, M.G.; Peclat, T.R.; Tchkonia, T.; Kirkland, J.L.; Chini, E. The NADase CD38 Is Induced by Factors Secreted from Senescent Cells Providing a Potential Link between Senescence and Age-Related Cellular NAD+ Decline. Biochem. Biophys. Res. Commun. 2019, 513, 486–493. [Google Scholar] [CrossRef]
- Graeff, R.; Liu, Q.; Kriksunov, I.A.; Hao, Q.; Lee, H.C. Acidic Residues at the Active Sites of CD38 and ADP-Ribosyl Cyclase Determine Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Synthesis and Hydrolysis Activities. J. Biol. Chem. 2006, 281, 28951–28957. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and Function of the ADP Ribosyl Cyclase/CD38 Gene Family in Physiology and Pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef]
- Ernst, I.M.A.; Fliegert, R.; Guse, A.H. Adenine Dinucleotide Second Messengers and T-Lymphocyte Calcium Signaling. Front. Immunol. 2013, 4, 259. [Google Scholar] [CrossRef]
- Yu, P.; Liu, Z.; Yu, X.; Ye, P.; Liu, H.; Xue, X.; Yang, L.; Li, Z.; Wu, Y.; Fang, C.; et al. Direct Gating of the TRPM2 Channel by cADPR via Specific Interactions with the ADPR Binding Pocket. Cell Rep. 2019, 27, 3684–3695.e4. [Google Scholar] [CrossRef]
- Gong, Y.-S.; Guo, J.; Hu, K.; Gao, Y.-Q.; Xie, B.-J.; Sun, Z.-D.; Yang, E.-N.; Hou, F.-L. Ameliorative Effect of Lotus Seedpod Proanthocyanidins on Cognitive Impairment and Brain Aging Induced by D-Galactose. Exp. Gerontol. 2016, 74, 21–28. [Google Scholar] [CrossRef]
- Reinders, M.E.J.; De Fijter, J.W.; Roelofs, H.; Bajema, I.M.; De Vries, D.K.; Schaapherder, A.F.; Claas, F.H.J.; Van Miert, P.P.M.C.; Roelen, D.L.; Van Kooten, C.; et al. Autologous Bone Marrow-Derived Mesenchymal Stromal Cells for the Treatment of Allograft Rejection After Renal Transplantation: Results of a Phase I Study. Stem Cells Transl. Med. 2013, 2, 107–111. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef]
- Chatterjee, S.; Daenthanasanmak, A.; Chakraborty, P.; Wyatt, M.W.; Dhar, P.; Selvam, S.P.; Fu, J.; Zhang, J.; Nguyen, H.; Kang, I.; et al. CD38-NAD+Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell Metab. 2018, 27, 85–100.e8. [Google Scholar] [CrossRef]
- Ortolan, E.; Augeri, S.; Fissolo, G.; Musso, I.; Funaro, A. CD157: From Immunoregulatory Protein to Potential Therapeutic Target. Immunol. Lett. 2019, 205, 59–64. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, S.; Liu, T.; Wang, H.; Liu, K.; Wang, Q.; Zeng, W. Sarm1/Myd88-5 Regulates Neuronal Intrinsic Immune Response to Traumatic Axonal Injuries. Cell Rep. 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Carty, M.; Goodbody, R.; Schröder, M.; Stack, J.; Moynagh, P.N.; Bowie, A.G. The Human Adaptor SARM Negatively Regulates Adaptor Protein TRIF-Dependent Toll-like Receptor Signaling. Nat. Immunol. 2006, 7, 1074–1081. [Google Scholar] [CrossRef]
- Panneerselvam, P.; Singh, L.P.; Selvarajan, V.; Chng, W.J.; Ng, S.B.; Tan, N.S.; Ho, B.; Chen, J.; Ding, J.L. T-Cell Death Following Immune Activation Is Mediated by Mitochondria-Localized SARM. Cell Death Differ. 2013, 20, 478–489. [Google Scholar] [CrossRef]
- Neukomm, L.J.; Burdett, T.C.; Seeds, A.M.; Hampel, S.; Coutinho-Budd, J.C.; Farley, J.E.; Wong, J.; Karadeniz, Y.B.; Osterloh, J.M.; Sheehan, A.E.; et al. Axon Death Pathways Converge on Axundead to Promote Functional and Structural Axon Disassembly. Neuron 2017, 95, 78–91.e5. [Google Scholar] [CrossRef]
- Berger, N.A. Poly(ADP-Ribose) in the Cellular Response to DNA Damage. Radiat. Res. 1985, 101, 4–15. [Google Scholar] [CrossRef]
- Li, J.-H.; Wang, Y.-H.; Zhu, X.-J.; Zhou, Q.; Xie, Z.-H.; Yao, T.-F. Metabolomics Study on the Association between Nicotinamide N-Methyltransferase Gene Polymorphisms and Type 2 Diabetes. Int. J. Diabetes Dev. Ctries 2018, 38, 409–416. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Chaverri, J. The Role of PI3K/AKT/mTOR Pathway in the Modulation of Autophagy and the Clearance of Protein Aggregates in Neurodegeneration. Cell Signal 2014, 26, 2694–2701. [Google Scholar] [CrossRef]
- Parsons, R.B.; Smith, M.-L.; Williams, A.C.; Waring, R.H.; Ramsden, D.B. Expression of Nicotinamide N-Methyltransferase (E.C. 2.1.1.1) in the Parkinsonian Brain. J. Neuropathol. Exp. Neurol. 2002, 61, 111–124. [Google Scholar] [CrossRef]
- Sazci, A.; Ozel, M.D.; Ergul, E.; Onder, M.E. Association of Nicotinamide-N-Methyltransferase (NNMT) Gene Rs694539 Variant with Bipolar Disorder. Gene 2013, 532, 272–275. [Google Scholar] [CrossRef]
- Bromberg, A.; Lerer, E.; Udawela, M.; Scarr, E.; Dean, B.; Belmaker, R.H.; Ebstein, R.; Agam, G. Nicotinamide-N-Methyltransferase (NNMT) in Schizophrenia: Genetic Association and Decreased Frontal Cortex mRNA Levels. Int. J. Neuropsychopharmacol. 2012, 15, 727–737. [Google Scholar] [CrossRef]
- Sazci, G.; Sazci, B.; Sazci, A.; Idrisoglu, H.A. Association of Nicotinamide-N-Methyltransferase Gene Rs694539 Variant with Epilepsy. Mol. Neurobiol. 2016, 53, 4197–4200. [Google Scholar] [CrossRef]
- Mateuszuk, L.; Jasztal, A.; Maslak, E.; Gasior-Glogowska, M.; Baranska, M.; Sitek, B.; Kostogrys, R.; Zakrzewska, A.; Kij, A.; Walczak, M.; et al. Antiatherosclerotic Effects of 1-Methylnicotinamide in Apolipoprotein E/Low-Density Lipoprotein Receptor-Deficient Mice: A Comparison with Nicotinic Acid. J. Pharmacol. Exp. Ther. 2016, 356, 514–524. [Google Scholar] [CrossRef]
- Yao, M.; Tabuchi, H.; Nagashima, Y.; Baba, M.; Nakaigawa, N.; Ishiguro, H.; Hamada, K.; Inayama, Y.; Kishida, T.; Hattori, K.; et al. Gene Expression Analysis of Renal Carcinoma: Adipose Differentiation-Related Protein as a Potential Diagnostic and Prognostic Biomarker for Clear-Cell Renal Carcinoma. J. Pathol. 2005, 205, 377–387. [Google Scholar] [CrossRef]
- Wu, Y.; Siadaty, M.S.; Berens, M.E.; Hampton, G.M.; Theodorescu, D. Overlapping Gene Expression Profiles of Cell Migration and Tumor Invasion in Human Bladder Cancer Identify Metallothionein 1E and Nicotinamide N-Methyltransferase as Novel Regulators of Cell Migration. Oncogene 2008, 27, 6679–6689. [Google Scholar] [CrossRef]
- Jang, J.S.; Cho, H.Y.; Lee, Y.J.; Ha, W.S.; Kim, H.W. The Differential Proteome Profile of Stomach Cancer: Identification of the Biomarker Candidates. Oncol. Res. 2004, 14, 491–499. [Google Scholar] [CrossRef]
- Roessler, M.; Rollinger, W.; Palme, S.; Hagmann, M.-L.; Berndt, P.; Engel, A.M.; Schneidinger, B.; Pfeffer, M.; Andres, H.; Karl, J.; et al. Identification of Nicotinamide N-Methyltransferase as a Novel Serum Tumor Marker for Colorectal Cancer. Clin. Cancer Res. 2005, 11, 6550–6557. [Google Scholar] [CrossRef]
- Sartini, D.; Santarelli, A.; Rossi, V.; Goteri, G.; Rubini, C.; Ciavarella, D.; Lo Muzio, L.; Emanuelli, M. Nicotinamide N-Methyltransferase Upregulation Inversely Correlates with Lymph Node Metastasis in Oral Squamous Cell Carcinoma. Mol. Med. 2007, 13, 415–421. [Google Scholar] [CrossRef]
- Tang, S.-W.; Yang, T.-C.; Lin, W.-C.; Chang, W.-H.; Wang, C.-C.; Lai, M.-K.; Lin, J.-Y. Nicotinamide N-Methyltransferase Induces Cellular Invasion through Activating Matrix Metalloproteinase-2 Expression in Clear Cell Renal Cell Carcinoma Cells. Carcinogenesis 2011, 32, 138–145. [Google Scholar] [CrossRef]
- Li, G.; Fang, S.; Shao, X.; Li, Y.; Tong, Q.; Kong, B.; Chen, L.; Wang, Y.; Yang, J.; Yu, H.; et al. Curcumin Reverses NNMT-Induced 5-Fluorouracil Resistance via Increasing ROS and Cell Cycle Arrest in Colorectal Cancer Cells. Biomolecules 2021, 11, 1295. [Google Scholar] [CrossRef]
- Tomida, M.; Ohtake, H.; Yokota, T.; Kobayashi, Y.; Kurosumi, M. Stat3 Up-Regulates Expression of Nicotinamide N-Methyltransferase in Human Cancer Cells. J. Cancer Res. Clin. Oncol. 2008, 134, 551–559. [Google Scholar] [CrossRef]
- Bose, S.; Allen, A.E.; Locasale, J.W. The Molecular Link from Diet to Cancer Cell Metabolism. Mol. Cell 2020, 80, 554. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Kim, J.; Hong, S.J.; Lim, E.K.; Yu, Y.-S.; Kim, S.W.; Roh, J.H.; Do, I.-G.; Joh, J.-W.; Kim, D.S. Expression of Nicotinamide N-Methyltransferase in Hepatocellular Carcinoma Is Associated with Poor Prognosis. J. Exp. Clin. Cancer Res. 2009, 28, 20. [Google Scholar] [CrossRef]
- Goldberg, R.B.; Mather, K. Targeting the Consequences of the Metabolic Syndrome in the Diabetes Prevention Program. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2077–2090. [Google Scholar] [CrossRef]
- Tripathi, B.K.; Srivastava, A.K. Diabetes Mellitus: Complications and Therapeutics. Med. Sci. Monit. 2006, 12, RA130–RA147. [Google Scholar]
- Yaguchi, H.; Togawa, K.; Moritani, M.; Itakura, M. Identification of Candidate Genes in the Type 2 Diabetes Modifier Locus Using Expression QTL. Genomics 2005, 85, 591–599. [Google Scholar] [CrossRef]
- Liu, M.; Chu, J.; Gu, Y.; Shi, H.; Zhang, R.; Wang, L.; Chen, J.; Shen, L.; Yu, P.; Chen, X.; et al. Serum N1-Methylnicotinamide Is Associated with Coronary Artery Disease in Chinese Patients. JAHA 2017, 6, e004328. [Google Scholar] [CrossRef]
- Liu, M.; He, A.; Chu, J.; Chen, C.; Zhang, S.; He, Y.; Tao, W.; Lu, M.; Hua, M.; Ju, W.; et al. Serum N1-Methylnicotinamide Is Associated with Left Ventricular Systolic Dysfunction in Chinese. Sci. Rep. 2018, 8, 8581. [Google Scholar] [CrossRef]
- Bubenek, S.; Nastase, A.; Niculescu, A.M.; Baila, S.; Herlea, V.; Lazar, V.; Paslaru, L.; Botezatu, A.; Tomescu, D.; Popescu, I.; et al. Assessment of Gene Expression Profiles in Peripheral Occlusive Arterial Disease. Can. J. Cardiol. 2012, 28, 712–720. [Google Scholar] [CrossRef]
- Herfindal, B.; Gerdts, E.; Kringeland, E.A.; Saeed, S.; Midtbø, H.; Halland, H. Concomitant Hypertension Is Associated with Abnormal Left Ventricular Geometry and Lower Systolic Myocardial Function in Overweight Participants: The FAT Associated CardiOvasculaR Dysfunction Study. J. Hypertens. 2020, 38, 1158–1164. [Google Scholar] [CrossRef]
- Kornev, M.; Caglayan, H.A.; Kudryavtsev, A.V.; Malyutina, S.; Ryabikov, A.; Schirmer, H.; Rösner, A. Influence of Hypertension on Systolic and Diastolic Left Ventricular Function Including Segmental Strain and Strain Rate. Echocardiography 2023, 40, 623–633. [Google Scholar] [CrossRef]
- Rus, R.R.; Pac, M.; Obrycki, Ł.; Sağsak, E.; Azukaitis, K.; Sinha, M.D.; Jankauskiene, A.; Litwin, M. Systolic and Diastolic Left Ventricular Function in Children with Primary Hypertension: A Systematic Review and Meta-Analysis. J. Hypertens. 2023, 41, 51–62. [Google Scholar] [CrossRef]
- Gao, Y.; Martin, N.I.; Van Haren, M.J. Nicotinamide N-Methyl Transferase (NNMT): An Emerging Therapeutic Target. Drug Discov. Today 2021, 26, 2699–2706. [Google Scholar] [CrossRef]
- Swaminathan, S.; Birudukota, S.; Thakur, M.K.; Parveen, R.; Kandan, S.; Juluri, S.; Shaik, S.; Anand, N.N.; Burri, R.R.; Kristam, R.; et al. Crystal Structures of Monkey and Mouse Nicotinamide N-Methyltransferase (NNMT) Bound with End Product, 1-Methyl Nicotinamide. Biochem. Biophys. Res. Commun. 2017, 491, 416–422. [Google Scholar] [CrossRef]
- Campagna, R.; Mateuszuk, Ł.; Wojnar-Lason, K.; Kaczara, P.; Tworzydło, A.; Kij, A.; Bujok, R.; Mlynarski, J.; Wang, Y.; Sartini, D.; et al. Nicotinamide N-Methyltransferase in Endothelium Protects against Oxidant Stress-Induced Endothelial Injury. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119082. [Google Scholar] [CrossRef]
- Dymkowska, D.; Wrzosek, A.; Zabłocki, K. Atorvastatin and Pravastatin Stimulate Nitric Oxide and Reactive Oxygen Species Generation, Affect Mitochondrial Network Architecture and Elevate Nicotinamide N-Methyltransferase Level in Endothelial Cells. J. Appl. Toxicol. 2021, 41, 1076–1088. [Google Scholar] [CrossRef]
- Zhou, Z.-Y.; Shi, W.-T.; Zhang, J.; Zhao, W.-R.; Xiao, Y.; Zhang, K.-Y.; Ma, J.; Tang, J.-Y.; Wang, Y. Sodium Tanshinone IIA Sulfonate Protects against Hyperhomocysteine-Induced Vascular Endothelial Injury via Activation of NNMT/SIRT1-Mediated NRF2/HO-1 and AKT/MAPKs Signaling in Human Umbilical Vascular Endothelial Cells. Biomed. Pharmacother. 2023, 158, 114137. [Google Scholar] [CrossRef]
- Watała, C.; Kaźmierczak, P.; Dobaczewski, M.; Przygodzki, T.; Bartuś, M.; Łomnicka, M.; Słomińska, E.M.; Durackova, Z.; Chłopicki, S. Anti-Diabetic Effects of 1-Methylnicotinamide (MNA) in Streptozocin-Induced Diabetes in Rats. Pharmacol. Rep. 2009, 61, 86–98. [Google Scholar] [CrossRef]
- Rygula, A.; Pacia, M.Z.; Mateuszuk, L.; Kaczor, A.; Kostogrys, R.B.; Chlopicki, S.; Baranska, M. Identification of a Biochemical Marker for Endothelial Dysfunction Using Raman Spectroscopy. Analyst 2015, 140, 2185–2189. [Google Scholar] [CrossRef]
- Chlopicki, S.; Swies, J.; Mogielnicki, A.; Buczko, W.; Bartus, M.; Lomnicka, M.; Adamus, J.; Gebicki, J. 1-Methylnicotinamide (MNA), a Primary Metabolite of Nicotinamide, Exerts Anti-Thrombotic Activity Mediated by a Cyclooxygenase-2/Prostacyclin Pathway. Br. J. Pharmacol. 2007, 152, 230–239. [Google Scholar] [CrossRef]
- Reiss, A.B.; Edelman, S.D. Recent Insights into the Role of Prostanoids in Atherosclerotic Vascular Disease. Curr. Vasc. Pharmacol. 2006, 4, 395–408. [Google Scholar] [CrossRef]
- Biedroń, R.; Ciszek, M.; Tokarczyk, M.; Bobek, M.; Kurnyta, M.; Słominska, E.M.; Smoleński, R.T.; Marcinkiewicz, J. 1-Methylnicotinamide and Nicotinamide: Two Related Anti-Inflammatory Agents That Differentially Affect the Functions of Activated Macrophages. Arch. Immunol. Ther. Exp. 2008, 56, 127–134. [Google Scholar] [CrossRef]
- Menon, R.M.; Adams, M.H.; González, M.A.; Tolbert, D.S.; Leu, J.H.; Cefali, E.A. Plasma and Urine Pharmacokinetics of Niacin and Its Metabolites from an Extended-Release Niacin Formulation. Int. J. Clin. Pharmacol. Ther. 2007, 45, 448–454. [Google Scholar] [CrossRef]
- Lukasova, M.; Hanson, J.; Tunaru, S.; Offermanns, S. Nicotinic Acid (Niacin): New Lipid-Independent Mechanisms of Action and Therapeutic Potentials. Trends Pharmacol. Sci. 2011, 32, 700–707. [Google Scholar] [CrossRef]
- Neelakantan, H.; Vance, V.; Wetzel, M.D.; Wang, H.-Y.L.; McHardy, S.F.; Finnerty, C.C.; Hommel, J.D.; Watowich, S.J. Selective and Membrane-Permeable Small Molecule Inhibitors of Nicotinamide N-Methyltransferase Reverse High Fat Diet-Induced Obesity in Mice. Biochem. Pharmacol. 2018, 147, 141–152. [Google Scholar] [CrossRef]
- Barrows, R.D.; Jeffries, D.E.; Vishe, M.; Tukachinsky, H.; Zheng, S.-L.; Li, F.; Ma, Z.; Li, X.; Jin, S.; Song, H.; et al. Potent Uncompetitive Inhibitors of Nicotinamide N-Methyltransferase (NNMT) as In Vivo Chemical Probes. J. Med. Chem. 2022, 65, 14642–14654. [Google Scholar] [CrossRef]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Sirtuins and NAD + in the Development and Treatment of Metabolic and Cardiovascular Diseases. Circ. Res. 2018, 123, 868–885. [Google Scholar] [CrossRef]
- Ungvari, Z.; Labinskyy, N.; Mukhopadhyay, P.; Pinto, J.T.; Bagi, Z.; Ballabh, P.; Zhang, C.; Pacher, P.; Csiszar, A. Resveratrol Attenuates Mitochondrial Oxidative Stress in Coronary Arterial Endothelial Cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1876–H1881. [Google Scholar] [CrossRef]
- Corbi, G.; Conti, V.; Troisi, J.; Colucci, A.; Manzo, V.; Di Pietro, P.; Calabrese, M.C.; Carrizzo, A.; Vecchione, C.; Ferrara, N.; et al. Cardiac Rehabilitation Increases SIRT1 Activity and β-Hydroxybutyrate Levels and Decreases Oxidative Stress in Patients with HF with Preserved Ejection Fraction. Oxidative Med. Cell. Longev. 2019, 2019, 7049237. [Google Scholar] [CrossRef]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone Bodies Mimic the Life Span Extending Properties of Caloric Restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef]
- Edwards, C.; Canfield, J.; Copes, N.; Rehan, M.; Lipps, D.; Bradshaw, P.C. D-Beta-Hydroxybutyrate Extends Lifespan in C. Elegans. Aging 2014, 6, 621–644. [Google Scholar] [CrossRef]
- Schmeisser, K.; Parker, J.A. Nicotinamide-N-Methyltransferase Controls Behavior, Neurodegeneration and Lifespan by Regulating Neuronal Autophagy. PLoS Genet. 2018, 14, e1007561. [Google Scholar] [CrossRef]
- Parsons, R.B.; Smith, S.W.; Waring, R.H.; Williams, A.C.; Ramsden, D.B. High Expression of Nicotinamide N-Methyltransferase in Patients with Idiopathic Parkinson’s Disease. Neurosci. Lett. 2003, 342, 13–16. [Google Scholar] [CrossRef]
- Milani, Z.H.; Ramsden, D.B.; Parsons, R.B. Neuroprotective Effects of Nicotinamide N-Methyltransferase and Its Metabolite 1-Methylnicotinamide. J. Biochem. Mol. Toxicol. 2013, 27, 451–456. [Google Scholar] [CrossRef]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and Tau: Two Convergence Points in Alzheimer’s Disease. J. Alzheimers Dis. 2013, 33 (Suppl. 1), S141–S144. [Google Scholar] [CrossRef]
- Thomas, M.G.; Saldanha, M.; Mistry, R.J.; Dexter, D.T.; Ramsden, D.B.; Parsons, R.B. Nicotinamide N-Methyltransferase Expression in SH-SY5Y Neuroblastoma and N27 Mesencephalic Neurones Induces Changes in Cell Morphology via Ephrin-B2 and Akt Signalling. Cell Death Dis. 2013, 4, e669. [Google Scholar] [CrossRef]
- Bhalla, S.; Mehan, S.; Khan, A.; Rehman, M.U. Protective Role of IGF-1 and GLP-1 Signaling Activation in Neurological Dysfunctions. Neurosci. Biobehav. Rev. 2022, 142, 104896. [Google Scholar] [CrossRef]
- Kannt, A.; Rajagopal, S.; Kadnur, S.V.; Suresh, J.; Bhamidipati, R.K.; Swaminathan, S.; Hallur, M.S.; Kristam, R.; Elvert, R.; Czech, J.; et al. A Small Molecule Inhibitor of Nicotinamide N-Methyltransferase for the Treatment of Metabolic Disorders. Sci. Rep. 2018, 8, 3660. [Google Scholar] [CrossRef]
- Doench, J.G.; Petersen, C.P.; Sharp, P.A. siRNAs Can Function as miRNAs. Genes. Dev. 2003, 17, 438–442. [Google Scholar] [CrossRef]
- Jackson, A.L.; Bartz, S.R.; Schelter, J.; Kobayashi, S.V.; Burchard, J.; Mao, M.; Li, B.; Cavet, G.; Linsley, P.S. Expression Profiling Reveals Off-Target Gene Regulation by RNAi. Nat. Biotechnol. 2003, 21, 635–637. [Google Scholar] [CrossRef]
- Jackson, A.L.; Burchard, J.; Schelter, J.; Chau, B.N.; Cleary, M.; Lim, L.; Linsley, P.S. Widespread siRNA “off-Target” Transcript Silencing Mediated by Seed Region Sequence Complementarity. RNA 2006, 12, 1179–1187. [Google Scholar] [CrossRef]
- Liang, G.; Li, Y.; Lin, Y.; Yang, X.; Yang, J.; Hu, S.; Liu, A. Nicotinamide N-Methyltransferase and Liver Diseases. Genes. Dis. 2023, 10, 1883–1893. [Google Scholar] [CrossRef]
- Chen, S.-H.; Yu, X. Human DNA Ligase IV Is Able to Use NAD+ as an Alternative Adenylation Donor for DNA Ends Ligation. Nucleic Acids Res. 2019, 47, 1321–1334. [Google Scholar] [CrossRef]
- Chłopicki, S.; Kurdziel, M.; Sternak, M.; Szafarz, M.; Szymura-Oleksiak, J.; Kamiński, K.; Żołądź, J.A. Single Bout of Endurance Exercise Increases NNMT Activity in the Liver and MNA Concentration in Plasma; the Role of IL-6. Pharmacol. Rep. 2012, 64, 369–376. [Google Scholar] [CrossRef]
- Li, J.-H.; Qiu, L.-Q.; Zhu, X.-J.; Cai, C.-X. Influence of Exercises Using Different Energy Metabolism Systems on NNMT Expression in Different Types of Skeletal Muscle Fibers. Sci. Sports 2017, 32, 27–32. [Google Scholar] [CrossRef]
- Zhou, Q.; Huang, Z.-G.; Zhu, X.-J.; Xie, Z.-H.; Yao, T.-F.; Wang, Y.-H.; Li, J.-H. Effects of Nicotinamide N-Methyltransferase (NNMT) Inhibition on the Aerobic and the Anaerobic Endurance Exercise Capacity. Sci. Sports 2018, 33, e159–e165. [Google Scholar] [CrossRef]
- Kim, H.C.; Mofarrahi, M.; Vassilakopoulos, T.; Maltais, F.; Sigala, I.; Debigare, R.; Bellenis, I.; Hussain, S.N.A. Expression and Functional Significance of Nicotinamide N-Methyl Transferase in Skeletal Muscles of Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2010, 181, 797–805. [Google Scholar] [CrossRef]

{kind=link}
| Type of Disease | Specific Diseases | Correlation | Reference |
|---|---|---|---|
| Diabetes | Type 2 diabetes | NNMT Active ↑; The rs1941404 variant of the NNMT gene is significantly associated with T2D | [165] |
| Neurodegenerative Diseases | Alzheimer’s disease | NNMT expression ↑ | [166] |
| Parkinson’s disease | [167] | ||
| Dementia | [27] | ||
| Attention deficit disorder | [28] | ||
| Bipolar disorder | The rs694539 variant of the NNMT gene is significantly associated with bipolar disorder | [168] | |
| Schizophrenia | The rs694539 variant of the NNMT gene is significantly associated with schizophrenia | [169] | |
| Epilepsy | The rs694539 variant of the NNMT gene is significantly associated with epilepsy | [170] | |
| Cancer | Bladder cancer | NNMT Active ↑; Migration, invasion, proliferation, and viability of cancer cells ↑ | [7,13,14] |
| Breast cancer | [7,13,14] | ||
| Colorectal cancer | [7,13,14] | ||
| Gastric cancer | [7,13,14] | ||
| Lung cancer | [7,13,14] | ||
| Oral cavity cancer | [7,13,14] | ||
| Ovarian cancer | [7,13,14] | ||
| Prostate cancers | [7,13,14] | ||
| Glioma cancer | [7,13,14] | ||
| Lymphomas | [7,13,14] | ||
| Insulinomas | [7,13,14] | ||
| Esophageal squamous carcinoma | NNMT Active ↑; survival of cancer cells in the chemotherapy setting ↑ | [110] | |
| Mesenchymal cancer | Overexpression of NNMT makes cancer cells resistant to chemoradiotherapy ↑ | [148] | |
| Cardiovascular Diseases | Hypertension | The rs1941404 variant of the NNMT gene is significantly associated with hypertension | [16] |
| Hyperlipidemia | The rs1941404 variant of the NNMT gene is significantly associated with hyperlipidemia | [17] | |
| Atherosclerosis | MNA, a product of NNMT, is effective at reducing atherosclerotic plaque formation | [171] | |
| Other Diseases | Sarcopenia | NNMT activates senescent muscle stem cells and improves the regeneration of ageing skeletal muscles | [10] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.-J.; Sun, W.-D.; Zhu, X.-J.; Mei, Y.-Z.; Li, W.-S.; Li, J.-H. Nicotinamide N-Methyltransferase (NNMT): A New Hope for Treating Aging and Age-Related Conditions. Metabolites 2024, 14, 343. https://doi.org/10.3390/metabo14060343
Li J-J, Sun W-D, Zhu X-J, Mei Y-Z, Li W-S, Li J-H. Nicotinamide N-Methyltransferase (NNMT): A New Hope for Treating Aging and Age-Related Conditions. Metabolites. 2024; 14(6):343. https://doi.org/10.3390/metabo14060343
Chicago/Turabian StyleLi, Jing-Jing, Wei-Dong Sun, Xiao-Juan Zhu, Ya-Zhong Mei, Wen-Song Li, and Jiang-Hua Li. 2024. "Nicotinamide N-Methyltransferase (NNMT): A New Hope for Treating Aging and Age-Related Conditions" Metabolites 14, no. 6: 343. https://doi.org/10.3390/metabo14060343
APA StyleLi, J.-J., Sun, W.-D., Zhu, X.-J., Mei, Y.-Z., Li, W.-S., & Li, J.-H. (2024). Nicotinamide N-Methyltransferase (NNMT): A New Hope for Treating Aging and Age-Related Conditions. Metabolites, 14(6), 343. https://doi.org/10.3390/metabo14060343