
Multimodal Mass Spectrometry Imaging of an Osteosarcoma Multicellular Tumour Spheroid Model to Investigate Drug-Induced Response

 , , , , and
, , , , and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Osteosarcoma 3D Cell Culture
2.2. Chemotherapeutic Agent Treatments
2.3. Microtissue Preparation
2.4. 3D Cell Viability Assay
2.5. Desorption Electrospray Ionisation-Mass Spectrometry Imaging (DESI-MSI)
2.6. Matrix-Assisted Laser Desorption Ionisation–Immunohistochemistry (MALDI-IHC)
2.7. Imaging Mass Cytometry by Time-of-Flight (IMC ToF)
2.8. Segmentation Analysis
2.9. Discriminatory Analysis
3. Results and Discussion
3.1. Characterisation of SAOS-2 MCTSs by DESI–MSI Revealed Tumour-Region-Specific Metabolism
3.2. Chemotherapeutic-Induced Response of OS MCTSs by DESI-MSI
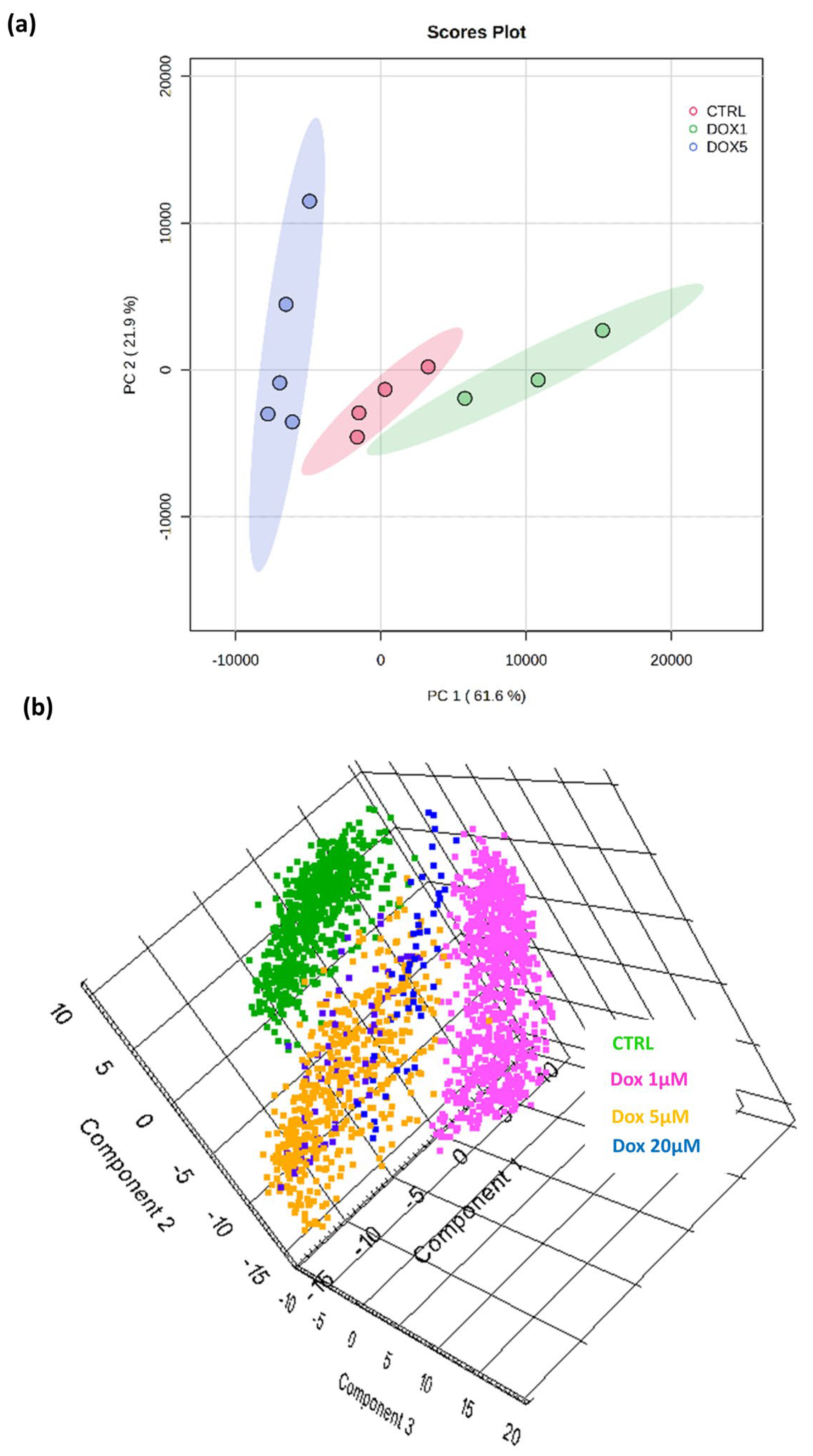
3.3. A Discriminatory Analysis of OS MCTSs Revealed Doxorubicin-Dose-Dependent Separation of DESI-MSI Data
3.4. Subcellular Protein Localisation Correlated with the Chemotherapeutic-Induced Metabolic Response of OS MCTSs by MALDI–IHC and IMC MSI
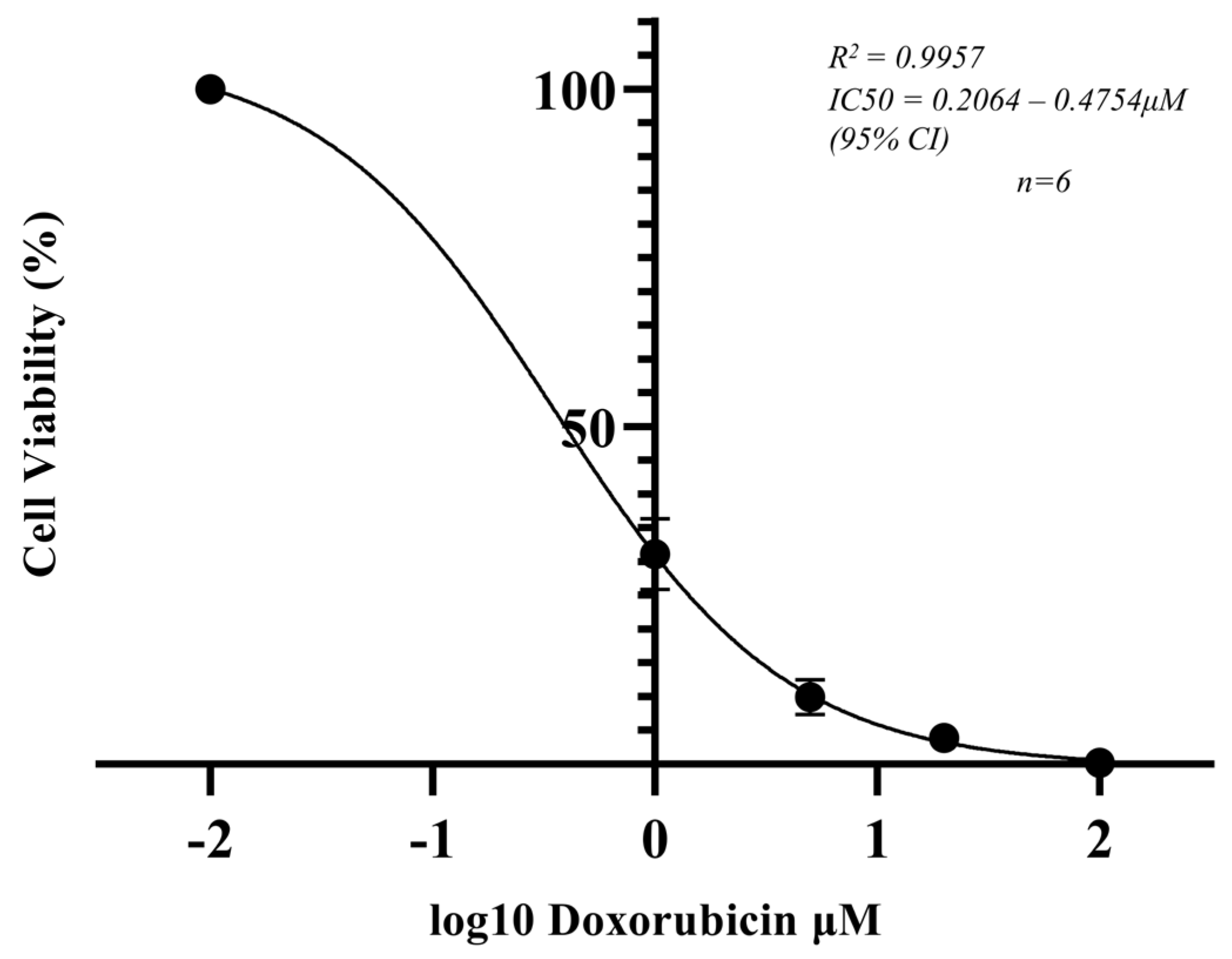
3.5. Cell Viability of OS MCTSs Determined by the ATP Concentration and Correlated with the Doxorubicin-Induced Metabolic Response
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van der Graaf, P.H. Probability of Success in Drug Development. Clin. Pharmacol. Ther. 2022, 111, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Fernando, K.; Menon, S.; Jansen, K.; Naik, P.; Nucci, G.; Roberts, J.; Wu, S.S.; Dolsten, M. Achieving end-to-end success in the clinic: Pfizer’s learnings on R&D productivity. Drug Discov. Today 2022, 27, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Eaton, B.R.; Schwarz, R.; Vatner, R.; Yeh, B.; Claude, L.; Indelicato, D.J.; Laack, N. Osteosarcoma. Pediatr. Blood Cancer 2021, 68, e28352. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, W.; Reininger-Gutmann, B.; Rinner, B.; Plasenzotti, R.; Wilflingseder, D.; De Kock, J.; Vanhaecke, T.; Rogiers, V.; Jírová, D.; Kejlová, K.; et al. The Current Status and Work of Three Rs Centres and Platforms in Europe. Altern. Lab. Anim. 2022, 50, 381–413. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, M.; Piccinini, F.; Arienti, C.; Zamagni, A.; Santi, S.; Polico, R.; Bevilacqua, A.; Tesei, A. 3D tumor spheroid models for in vitro therapeutic screening: A systematic approach to enhance the biological relevance of data obtained. Sci. Rep. 2016, 6, 19103. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of Multicell Spheroids in Tissue Culture as a Model of Nodular Carcinomas. J. Natl. Cancer Inst. 1971, 46, 113–120. [Google Scholar] [CrossRef]
- Palubeckaitė, I.; Crooks, L.; Smith, D.P.; Cole, L.M.; Bram, H.; Le Maitre, C.; Clench, M.R.; Cross, N.A. Mass spectrometry imaging of endogenous metabolites in response to doxorubicin in a novel 3D osteosarcoma cell culture model. J. Mass Spectrom. 2020, 55, e4461. [Google Scholar] [CrossRef]
- Flint, L.E.; Hamm, G.R.; Ready, J.D.; Ling, S.; Duckett, C.J.; Cross, N.A.; Cole, L.M.; Smith, D.P.; Goodwin, R.J.A.; Clench, M.R. Characterization of an Aggregated Three-Dimensional Cell Culture Model by Multimodal Mass Spectrometry Imaging. Anal. Chem. 2020, 92, 12538–12547. [Google Scholar] [CrossRef]
- Flint, L.E.; Hamm, G.; Ready, J.D.; Ling, S.; Duckett, C.J.; Cross, N.A.; Cole, L.M.; Smith, D.P.; Goodwin, R.J.A.; Clench, M.R. Comparison of osteosarcoma aggregated tumour models with human tissue by multimodal mass spectrometry imaging. Metabolites 2021, 11, 506. [Google Scholar] [CrossRef]
- Cooper-Shepherd, D.A.; Wildgoose, J.; Kozlov, B.; Johnson, W.J.; Tyldesley-Worster, R.; Palmer, M.E.; Hoyes, J.B.; McCullagh, M.; Jones, E.; Tonge, R.; et al. Novel Hybrid Quadrupole-Multireflecting Time-of-Flight Mass Spectrometry System. J. Am. Soc. Mass Spectrom. 2023, 34, 264–272. [Google Scholar] [CrossRef]
- Takaáts, Z.; Wiseman, J.M.; Gologan, B.; Cooks, R.G. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science 2004, 306, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Ifa, D.R.; Wiseman, J.M.; Song, Q.; Cooks, R.G. Development of capabilities for imaging mass spectrometry under ambient conditions with desorption electrospray ionization (DESI). Int. J. Mass Spectrom. 2007, 259, 8–15. [Google Scholar] [CrossRef]
- Yagnik, G.; Liu, Z.; Rothschild, K.J.; Lim, M.J. Highly Multiplexed Immunohistochemical MALDI-MS Imaging of Biomarkers in Tissues. J. Am. Soc. Mass Spectrom. 2021, 32, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Claes, B.S.R.; Krestensen, K.K.; Yagnik, G.; Grgic, A.; Kuik, C.; Lim, M.J.; Rothschild, K.J.; Vandenbosch, M.; Heeren, R.M.A. MALDI-IHC-Guided In-Depth Spatial Proteomics: Targeted and Untargeted MSI Combined. Anal. Chem. 2023, 95, 2329–2338. [Google Scholar] [CrossRef] [PubMed]
- Stack, E.C.; Wang, C.; Roman, K.A.; Hoyt, C.C. Multiplexed immunohistochemistry, imaging, and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 2014, 70, 46–58. [Google Scholar] [CrossRef]
- Rathore, R.; Van Tine, B.A. Pathogenesis and Current Treatment of Osteosarcoma: Perspectives for Future Therapies. J. Clin. Med. 2021, 10, 1182. [Google Scholar] [CrossRef]
- Link, M.P.; Goorin, A.M.; Horowitz, M.; Meyer, W.H.; Belasco, J.; Baker, A.; Ayala, A.; Shuster, J. Adjuvant chemotherapy of high-grade osteosarcoma of the extremity. Updated results of the Multi-Institutional Osteosarcoma Study. Clin. Orthop. Relat. Res. 1991, 270, 8–14. [Google Scholar] [CrossRef]
- Isakoff, M.S.; Bielack, S.S.; Meltzer, P.; Gorlick, R. Osteosarcoma: Current Treatment and a Collaborative Pathway to Success. J. Clin. Oncol. 2015, 33, 3029–3035. [Google Scholar] [CrossRef]
- Arcamone, F.; Cassinelli, G.; Fantini, G.; Grein, A.; Orezzi, P.; Pol, C.; Spalla, C. Adriamycin, 14-hydroxydaimomycin, a new antitumor antibiotic from S. Peucetius var. caesius. Biotechnol. Bioeng. 1969, 11, 1101–1110. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Kołat, D.; Kałuzińska-Kołat, Ż.; Celik, I.; Kontek, R. Doxorubicin—An Agent with Multiple Mechanisms of Anticancer Activity. Cells 2023, 12, 659. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Marchandet, L.; Lallier, M.; Charrier, C.; Baud’huin, M.; Ory, B.; Lamoureux, F. Mechanisms of Resistance to Conventional Therapies for Osteosarcoma. Cancers 2021, 13, 683. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.J.; Fojo, A.T.; Ueda, K.; Crist, W.; Green, A.; Brodeur, G.; Pastan, I.; Gottesman, M.M. Expression of the multidrug resistance, MDR1, gene in neuroblastomas. J. Clin. Oncol. 1990, 8, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Mahon, F.-X.; Belloc, F.; Lagarde, V.; Chollet, C.; Moreau-Gaudry, F.; Reiffers, J.; Goldman, J.M.; Melo, J.V. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood 2003, 101, 2368–2373. [Google Scholar] [CrossRef]
- Belisario, D.C.; Akman, M.; Godel, M.; Campani, V.; Patrizio, M.P.; Scotti, L.; Hattinger, C.M.; De Rosa, G.; Donadelli, M.; Serra, M.; et al. ABCA1/ABCB1 Ratio Determines Chemo- and Immune-Sensitivity in Human Osteosarcoma. Cells 2020, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Khanna, C.; Prehn, J.; Yeung, C.; Caylor, J.; Tsokos, M.; Helman, L. An orthotopic model of murine osteosarcoma with clonally related variants differing in pulmonary metastatic potential. Clin. Exp. Metastasis 2000, 18, 261–271. [Google Scholar] [CrossRef]
- Ren, L.; Hong, E.S.; Mendoza, A.; Issaq, S.; Hoang, C.T.; Lizardo, M.; LeBlanc, A.; Khanna, C. Metabolomics uncovers a link between inositol metabolism and osteosarcoma metastasis. Oncotarget 2017, 8, 38541–38553. [Google Scholar] [CrossRef]
- Fan, T.M.; Roberts, R.D.; Lizardo, M.M. Understanding and Modeling Metastasis Biology to Improve Therapeutic Strategies for Combating Osteosarcoma Progression. Front. Oncol. 2020, 10, 13. [Google Scholar] [CrossRef]
- Freyer, J.P.; Sutherland, R.M. Selective dissociation and characterization of cells from different regions of multicell tumor spheroids. Cancer Res. 1980, 40, 3956–3965. [Google Scholar]
- Dannhorn, A.; Kazanc, E.; Ling, S.; Nikula, C.; Karali, E.; Serra, M.P.; Vorng, J.-L.; Inglese, P.; Maglennon, G.; Hamm, G.R.; et al. Universal Sample Preparation Unlocking Multimodal Molecular Tissue Imaging. Anal. Chem. 2020, 92, 11080–11088. [Google Scholar] [CrossRef]
- Swales, J.G.; Dexter, A.; Hamm, G.; Nilsson, A.; Strittmatter, N.; Michopoulos, F.; Hardy, C.; Morentin-Gutierrez, P.; Mellor, M.; Andren, P.E.; et al. Quantitation of Endogenous Metabolites in Mouse Tumors Using Mass-Spectrometry Imaging. Anal. Chem. 2018, 90, 6051–6058. [Google Scholar] [CrossRef]
- Promega Corporation. CellTiter-Glo® 3D Cell Viability Assay 3D Cell Culture. Available online: https://www.promega.co.uk/products/cell-health-assays/cell-viability-and-cytotoxicity-assays/celltiter-glo-3d-cell-viability-assay/?catNum=G9681 (accessed on 12 October 2023).
- Race, A.M.; Styles, I.B.; Bunch, J. Inclusive sharing of mass spectrometry imaging data requires a converter for all. J. Proteom. 2012, 75, 5111–5112. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.-L.; et al. Fatty Acid Uptake and Lipid Storage Induced by HIF-1α Contribute to Cell Growth and Survival after Hypoxia-Reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef]
- Xu, S.; Jay, A.; Brunaldi, K.; Huang, N.; Hamilton, J.A. CD36 enhances fatty acid uptake by increasing the rate of intracellular esterification but not transport across the plasma membrane. Biochemistry 2013, 52, 7254–7261. [Google Scholar] [CrossRef] [PubMed]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Too complex to fail? Targeting fatty acid metabolism for cancer therapy. Prog. Lipid Res. 2022, 85, 101143. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.-H.; Su, Y.-C.; Lai, P.-L.; Zhang, Y.; Xu, Y.-F.; Zhao, A.; Yao, G.-Y.; Jia, C.-H.; Lin, J.; Xu, S.; et al. Critical role of arachidonic acid-activated mTOR signaling in breast carcinogenesis and angiogenesis. Oncogene 2013, 32, 160–170. [Google Scholar] [CrossRef]
- Alexander, L.D.; Cui, X.-L.; Falck, J.R.; Douglas, J.G. Arachidonic acid directly activates members of the mitogen-activated protein kinase superfamily in rabbit proximal tubule cells. Kidney Int. 2001, 59, 2039–2053. [Google Scholar] [CrossRef]
- Garcia, M.C.; Ray, D.M.; Lackford, B.; Rubino, M.; Olden, K.; Roberts, J.D. Arachidonic Acid Stimulates Cell Adhesion through a Novel p38 MAPK-RhoA Signaling Pathway That Involves Heat Shock Protein 27. J. Biol. Chem. 2009, 284, 20936–20945. [Google Scholar] [CrossRef] [PubMed]
- Hughes-Fulford, M.; Li, C.-F.; Boonyaratanakornkit, J.; Sayyah, S. Arachidonic acid activates phosphatidylinositol 3-kinase signaling and induces gene expression in prostate cancer. Cancer Res. 2006, 66, 1427–1433. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Karali, E.; Tripp, A.; Valle, A.; Inglese, P.; Perry, N.J.; Magee, D.J.; Virmouni, S.A.; Elder, G.A.; Tyson, A.L.; et al. Metabolic Fingerprinting Links Oncogenic PIK3CA with Enhanced Arachidonic Acid-Derived Eicosanoids. Cell 2020, 181, 1596–1611.e27. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Sun, J.; Wang, H.; Zhou, C.; Li, C.; Wu, Y.; Wen, Y.; Zhang, X.; Ren, X.; Guo, Z.; et al. Far upstream element-binding protein 1 confers lobaplatin resistance by transcriptionally activating PTGES and facilitating the arachidonic acid metabolic pathway in osteosarcoma. MedComm 2023, 4, e257. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Dufour, M.; Faes, S.; Dormond-Meuwly, A.; Demartines, N.; Dormond, O. PGE2-induced colon cancer growth is mediated by mTORC1. Biochem. Biophys. Res. Commun. 2014, 451, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xu, Y.; Brooks, A.; Guo, B.; Miskimins, K.W.; Qian, S.Y. Knockdown delta-5-desaturase promotes the formation of a novel free radical byproduct from COX-catalyzed ω-6 peroxidation to induce apoptosis and sensitize pancreatic cancer cells to chemotherapy drugs. Free Radic. Biol. Med. 2016, 97, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Kubota, N.; Ozawa, F.; Okada, S.; Inada, T.; Komatsu, K.; Okayasu, R. The phosphatidylinositol 3-kinase inhibitor wortmannin sensitizes quiescent but not proliferating MG-63 human osteosarcoma cells to radiation. Cancer Lett. 1998, 133, 161–167. [Google Scholar] [CrossRef]
- Tata, A.; Woolman, M.; Ventura, M.; Bernards, N.; Ganguly, M.; Gribble, A.; Shrestha, B.; Bluemke, E.; Ginsberg, H.J.; Vitkin, A.; et al. Rapid Detection of Necrosis in Breast Cancer with Desorption Electrospray Ionization Mass Spectrometry. Sci. Rep. 2016, 6, 35374. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, K.W.; Gray, R.; Fowble, B.; Tormey, D.C.; Taylor, S.G. Tumor necrosis is a prognostic predictor for early recurrence and death in lymph node-positive breast cancer: A 10-year follow-up study of 728 Eastern Cooperative Oncology Group patients. J. Clin. Oncol. 1993, 11, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Swinson, D.E.; Jones, J.; Richardson, D.; Cox, G.; Edwards, J.G.; O’Byrne, K.J. Tumour necrosis is an independent prognostic marker in non-small cell lung cancer: Correlation with biological variables. Lung Cancer 2002, 37, 235–240. [Google Scholar] [CrossRef]
- Edwards, J.G.; Swinson, D.E.; Jones, J.L.; Muller, S.; Waller, D.A.; O’byrne, K.J. Tumor Necrosis Correlates with Angiogenesis and Is a Predictor of Poor Prognosis in Malignant Mesothelioma. Chest 2003, 124, 1916–1923. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ma, Z.; Wang, Y.; Wang, Z.; Tian, Y.; Du, Y.; Bian, W.; Duan, Y.; Liu, J. Necrosis of osteosarcoma cells induces the production and release of high-mobility group box 1 protein. Exp. Ther. Med. 2018, 15, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ni, J.; Liu, K.; Yu, Y.; Xie, M.; Kang, R.; Vernon, P.; Cao, L.; Tang, D. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res. 2012, 72, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ashana, A.O.; Moretti, V.M.; Lackman, R.D. The relation of tumour necrosis and survival in patients with osteosarcoma. Int. Orthop. 2011, 35, 1847–1853. [Google Scholar] [CrossRef] [PubMed]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and Thioredoxin Antioxidant Pathways Synergize to Drive Cancer Initiation and Progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef]
- Bachhawat, A.K.; Yadav, S. The glutathione cycle: Glutathione metabolism beyond the γ-glutamyl cycle. IUBMB Life 2018, 70, 585–592. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Fu, Y.; Zou, T.; Shen, X.; Nelson, P.J.; Li, J.; Wu, C.; Yang, J.; Zheng, Y.; Bruns, C.; Zhao, Y.; et al. Lipid metabolism in cancer progression and therapeutic strategies. MedComm 2021, 2, 27–59. [Google Scholar] [CrossRef]
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.-Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty Acid Synthase Gene Is Up-regulated by Hypoxia via Activation of Akt and Sterol Regulatory Element Binding Protein-1. Cancer Res. 2008, 68, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Mentoor, I.; Nell, T.; Emjedi, Z.; van Jaarsveld, P.J.; de Jager, L.; Engelbrecht, A.-M. Decreased Efficacy of Doxorubicin Corresponds with Modifications in Lipid Metabolism Markers and Fatty Acid Profiles in Breast Tumors from Obese vs. Lean Mice. Front. Oncol. 2020, 10, 306. [Google Scholar] [CrossRef]
- Kreuzaler, P.; Inglese, P.; Ghanate, A.; Gjelaj, E.; Wu, V.; Panina, Y.; Mendez-Lucas, A.; MacLachlan, C.; Patani, N.; Hubert, C.B.; et al. Vitamin B5 supports MYC oncogenic metabolism and tumor progression in breast cancer. Nat. Metab. 2023, 5, 1870–1886. [Google Scholar] [CrossRef]
- Heselmeyer-Haddad, K.; Garcia, L.Y.B.; Bradley, A.; Ortiz-Melendez, C.; Lee, W.-J.; Christensen, R.; Prindiville, S.A.; Calzone, K.A.; Soballe, P.W.; Hu, Y.; et al. Single-Cell Genetic Analysis of Ductal Carcinoma in Situ and Invasive Breast Cancer Reveals Enormous Tumor Heterogeneity yet Conserved Genomic Imbalances and Gain of MYC during Progression. Am. J. Pathol. 2012, 181, 1807–1822. [Google Scholar] [CrossRef]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef]
- Horbay, R.; Hamraghani, A.; Ermini, L.; Holcik, S.; Beug, S.T.; Yeganeh, B. Role of Ceramides and Lysosomes in Extracellular Vesicle Biogenesis, Cargo Sorting and Release. Int. J. Mol. Sci. 2022, 23, 15317. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Endl, E.; Gerdes, J. The Ki-67 Protein: Fascinating Forms and an Unknown Function. Exp. Cell Res. 2000, 257, 231–237. [Google Scholar] [CrossRef]
- Airley, R.; Loncaster, J.; Davidson, S.; Bromley, M.; Roberts, S.; Patterson, A.; Hunter, R.; Stratford, I.; West, C. Glucose Transporter Glut-1 Expression Correlates with Tumor Hypoxia and Predicts Metastasis-Free Survival in Advanced Carcinoma of the Cervix. Clin. Cancer Res. 2001, 7, 928–934. [Google Scholar]
- Mutsaers, A.J.; Walkley, C.R. Cells of origin in osteosarcoma: Mesenchymal stem cells or osteoblast committed cells? Bone 2014, 62, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Berr, A.L.; Wiese, K.; dos Santos, G.; Koch, C.M.; Anekalla, K.R.; Kidd, M.; Davis, J.M.; Cheng, Y.; Hu, Y.-S.; Ridge, K.M. Vimentin is required for tumor progression and metastasis in a mouse model of non-small cell lung cancer. Oncogene 2023, 42, 2074–2087. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, S.; Azad, G.K.; Amen, T.; Brielle, S.; Park, J.E.; Sze, S.K.; Meshorer, E.; Kaganovich, D. Vimentin protects differentiating stem cells from stress. Sci. Rep. 2020, 10, 19525. [Google Scholar] [CrossRef]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [PubMed]
- Seip, K.; Fleten, K.G.; Barkovskaya, A.; Nygaard, V.; Haugen, M.H.; Engesæter, B.; Mælandsmo, G.M.; Prasmickaite, L. Fibroblast-induced switching to the mesenchymal-like phenotype and PI3K/mTOR signaling protects melanoma cells from BRAF inhibitors. Oncotarget 2016, 7, 19997–20015. [Google Scholar] [CrossRef] [PubMed]
- McCain, J. The MAPK (ERK) Pathway: Investigational Combinations for the Treatment of BRAF-Mutated Metastatic Melanoma. Pharm. Ther. 2013, 38, 96–108. [Google Scholar]
- Rupp, T.; Langlois, B.; Koczorowska, M.M.; Radwanska, A.; Sun, Z.; Hussenet, T.; Lefebvre, O.; Murdamoothoo, D.; Arnold, C.; Klein, A.; et al. Tenascin-C Orchestrates Glioblastoma Angiogenesis by Modulation of Pro- and Anti-angiogenic Signaling. Cell Rep. 2016, 17, 2607–2619. [Google Scholar] [CrossRef]
- Hasim, M.S.; Nessim, C.; Villeneuve, P.J.; Vanderhyden, B.C.; Dimitroulakos, J. Activating Transcription Factor 3 as a Novel Regulator of Chemotherapy Response in Breast Cancer. Transl. Oncol. 2018, 11, 988–998. [Google Scholar] [CrossRef]
- Park, E.-J.; Kwon, H.-K.; Choi, Y.-M.; Shin, H.-J.; Choi, S. Doxorubicin Induces Cytotoxicity through Upregulation of pERK–Dependent ATF3. PLoS ONE 2012, 7, e44990. [Google Scholar] [CrossRef]
- Riss, T.; Valley, M.; Kupcho, K.; Zimprich, C.; Leippe, D.; Niles, A.; Vidugiriene, J.; Cali, J.; Kelm, J.; Moritz, W.; et al. Validation of In Vitro Assays to Measure Cytotoxicity in 3D Cell Cultures. Toxicol. Lett. 2014, 229, S145. [Google Scholar] [CrossRef]
- Khalidi, H.; Onasanwo, A.; Islam, B.; Jo, H.; Fisher, C.; Aidley, R.; Gardner, I.; Bois, F.Y. SimRFlow: An R-based workflow for automated high-throughput PBPK simulation with the Simcyp® simulator. Front. Pharmacol. 2022, 13, 929200. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal | Antibody Target | Clone | [Conjugated Ab] |
|---|---|---|---|
| 143Nd | Vimentin | RV202 | 1:200 |
| 160Gd | GLUT1 | EPR3915 | 1:100 |
| 166Er | EpCam | G8.8 | 1:100 |
| 167Er | Tenascin C | AZCYT0346/Poly | 1:100 |
| 168Er | Ki67 | B56 | 1:100 |
| 171Yb | ATPase | EP1845Y/AZCYT0253 | 1:100 |
| 175Lu | pS6 | N7-548 | 1:250 |
| 173Yb | γH2AX | JBW301 | 1:50 |
| 176Lu | pHH3 | HTA28 | 1:200 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pearce, S.M.; Cross, N.A.; Smith, D.P.; Clench, M.R.; Flint, L.E.; Hamm, G.; Goodwin, R.; Langridge, J.I.; Claude, E.; Cole, L.M. Multimodal Mass Spectrometry Imaging of an Osteosarcoma Multicellular Tumour Spheroid Model to Investigate Drug-Induced Response. Metabolites 2024, 14, 315. https://doi.org/10.3390/metabo14060315
Pearce SM, Cross NA, Smith DP, Clench MR, Flint LE, Hamm G, Goodwin R, Langridge JI, Claude E, Cole LM. Multimodal Mass Spectrometry Imaging of an Osteosarcoma Multicellular Tumour Spheroid Model to Investigate Drug-Induced Response. Metabolites. 2024; 14(6):315. https://doi.org/10.3390/metabo14060315
Chicago/Turabian StylePearce, Sophie M., Neil A. Cross, David P. Smith, Malcolm R. Clench, Lucy E. Flint, Gregory Hamm, Richard Goodwin, James I. Langridge, Emmanuelle Claude, and Laura M. Cole. 2024. "Multimodal Mass Spectrometry Imaging of an Osteosarcoma Multicellular Tumour Spheroid Model to Investigate Drug-Induced Response" Metabolites 14, no. 6: 315. https://doi.org/10.3390/metabo14060315
APA StylePearce, S. M., Cross, N. A., Smith, D. P., Clench, M. R., Flint, L. E., Hamm, G., Goodwin, R., Langridge, J. I., Claude, E., & Cole, L. M. (2024). Multimodal Mass Spectrometry Imaging of an Osteosarcoma Multicellular Tumour Spheroid Model to Investigate Drug-Induced Response. Metabolites, 14(6), 315. https://doi.org/10.3390/metabo14060315