
Elucidating the Role of Lipid-Metabolism-Related Signal Transduction and Inhibitors in Skin Cancer



Abstract
1. Introduction
2. Signaling Pathways and Target Proteins Associated with Lipid Metabolism in Skin Cancer
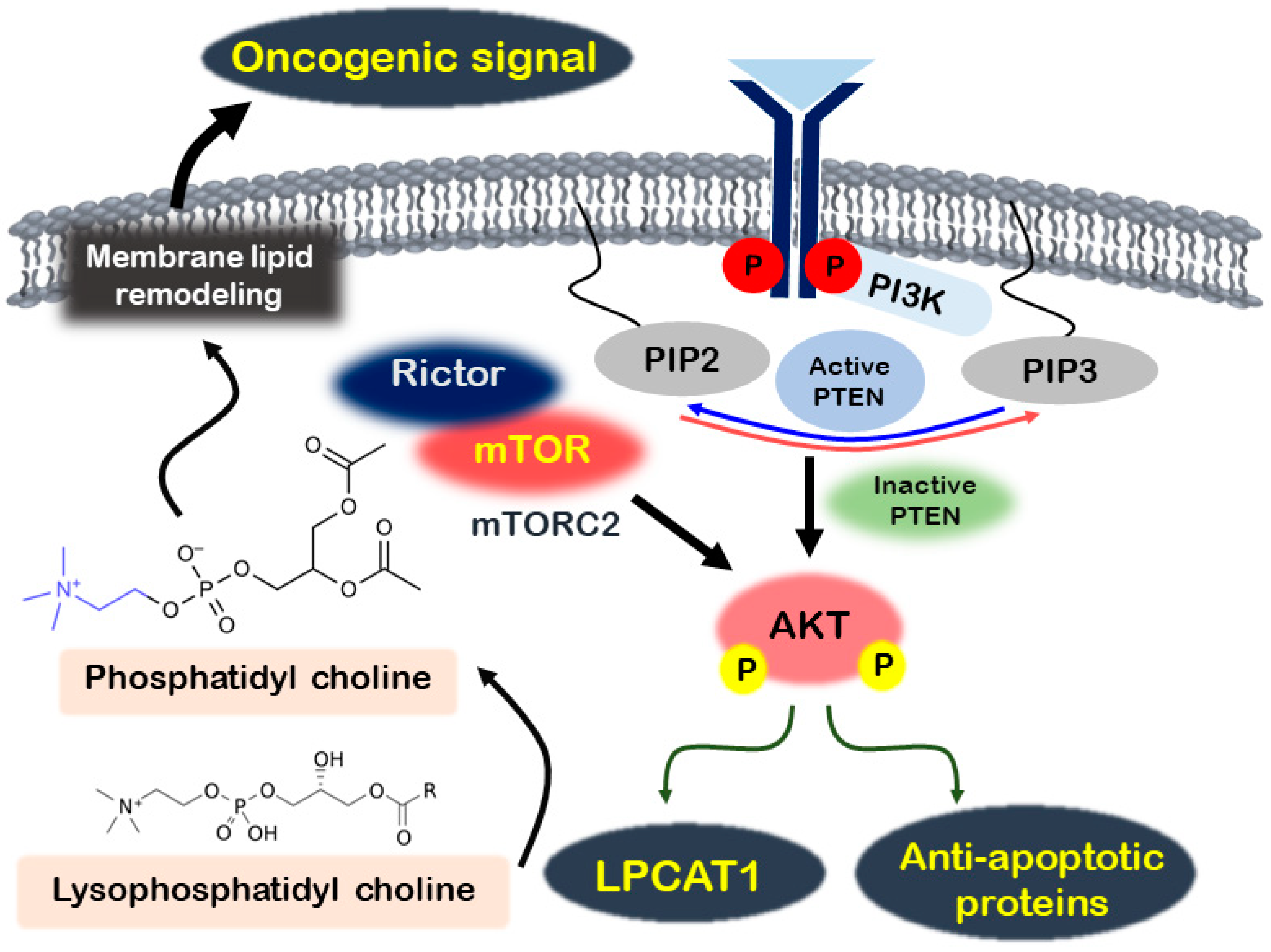
2.1. PI3K/AKT/mTOR Signaling
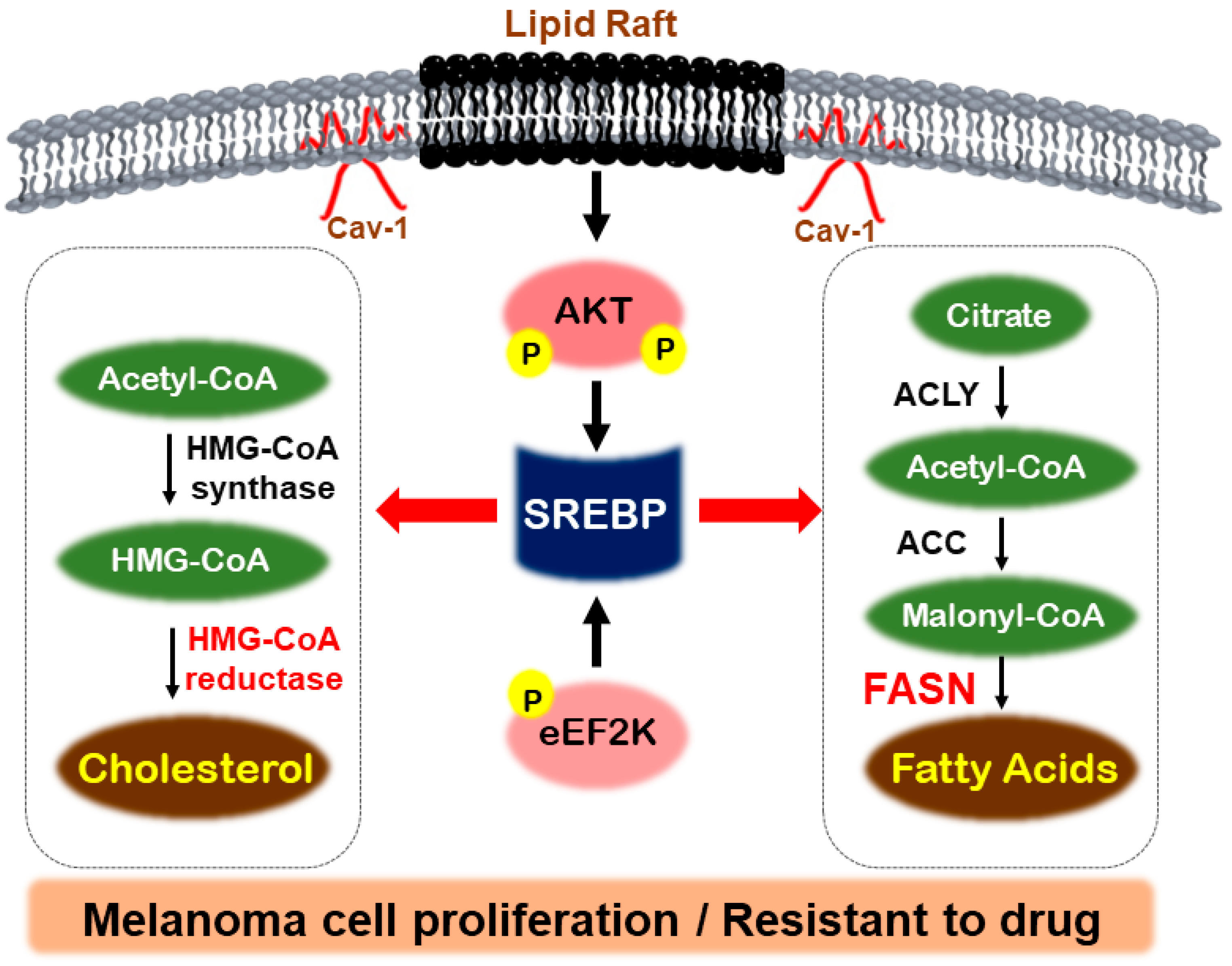
2.2. Fatty Acid Synthase (FASN)
2.3. Sterol Regulatory-Element-Binding Proteins (SREBPs)
2.4. Peroxisome Proliferator-Activated Receptor-Gamma Coactivator (PGC)-1α

{kind=link}
{kind=link}
| Target Proteins | Effects on Melanoma Cells | Reference |
|---|---|---|
| PI3Kδ ↓ | IL-22-induced psoriatic keratinocyte proliferation ↓ | [35] |
| mTOR ↓ | Invasiveness of melanoma cells in organotypic cultures ↓ | [35] |
| LPCAT1 ↑ | Akt-dependent cell growth ↑ | [43] |
| PTEN ↓ | Proliferation of melanoma cells through FRA1 expression ↓ | [44] |
| FASN ↑ | Lipogenesis in B-Raf mutant patient-derived xenograft ↑ | [51] |
| FASN ↓ | Invasiveness through VEGFA165b production ↓ | [55] |
| FASN ↓ | Proliferation of melanoma cells through Cav-1 palmitoylation ↓ | [56] |
| DHCR24 ↓ | Sensitivity of melanoma cells against oxidative stress ↑ | [52] |
| Ganglioside GD3 ↑ | Growth and invasion through SREBP activation ↑ | [52] |
| PGC-1α ↓ | Metastasis through integrin expression ↑ | [71] |
| PGC-1α ↑ | Mitochondrial energy metabolism and ROS detoxification ↑ | [74] |
3. Therapeutic Inhibitors Targeting Lipid Metabolism in Melanoma
3.1. Orlistat
3.2. Cerulenin
3.3. TVB-3644
3.4. Fatostatin
3.5. Betulin/Betulinic Acid
| Small-Molecule Inhibitors | Molecular Targets | Effects on Melanoma Cells | Ref. |
|---|---|---|---|
| Orlistat |
|
| [76,77,78,80] |
| Cerulenin C75 |
|
| [77,78,79,81] |
| TVB-3644 |
|
| [51,83]. |
| Fatostatin |
|
| [85,86]. |
| Betulin/Betulinic acid |
|
| [85,89] |
4. Lipid Metabolic Dynamics in the Melanoma Microenvironment
5. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Cavaco, M.C.; Pereira, C.; Kreutzer, B.; Gouveia, L.F.; Silva-Lima, B.; Brito, A.M.; Videira, M. Evading P-glycoprotein mediated-efflux chemoresistance using solid lipid nanoparticles. Eur. J. Pharm. Biopharm. 2017, 110, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Xu, Z.; Huang, Z.; Tang, Y.; Yang, D.; Huang, J.; He, L.; Liu, M.; Chen, Z.; Teng, Y. CPI-613 rewires lipid metabolism to enhance pancreatic cancer apoptosis via the AMPK-ACC signaling. J. Exp. Clin. Cancer Res. 2020, 39, 73. [Google Scholar] [CrossRef]
- Khan, N.R.; Wong, T.W. 5-Fluorouracil ethosomes-skin deposition and melanoma permeation synergism with microwave. Artif. Cells Nanomed. Biotechnol. 2018, 46, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Alicea, G.M.; Rebecca, V.W.; Goldman, A.R.; Fane, M.E.; Douglass, S.M.; Behera, R.; Webster, M.R.; Kugel, C.H., 3rd; Ecker, B.L.; Caino, M.C.; et al. Changes in aged fibroblast lipid metabolism induce age-dependent melanoma cell resistance to targeted therapy via the fatty acid transporter FATP2. Cancer Discov. 2020, 10, 1282–1295. [Google Scholar] [CrossRef]
- Qian, L.; Wang, G.; Li, B.; Su, H.; Qin, L. Regulation of lipid metabolism by APOE4 in intrahepatic cholangiocarcinoma via the enhancement of ABCA1 membrane expression. PeerJ 2024, 12, e16740. [Google Scholar] [CrossRef]
- Holloway, J.; Seeley, A.; Cobbe, N.; Turkington, R.C.; Longley, D.B.; Evergren, E. The E3 ubiquitin ligase Itch regulates death receptor and cholesterol trafficking to affect TRAIL-mediated apoptosis. Cell Death Dis. 2024, 15, 40. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, P.; Xu, J.; Lv, G.; Li, Y. Lipid metabolism in tumor microenvironment: Novel therapeutic targets. Cancer Cell Int. 2022, 22, 224. [Google Scholar] [CrossRef]
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tait-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A stromal lysolipid-autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discov. 2019, 9, 617–627. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Ha, J.H.; Jayaraman, M.; Liu, J.; Moxley, K.M.; Isidoro, C.; Sood, A.K.; Song, Y.S.; Dhanasekaran, D.N. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett. 2019, 442, 464–474. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Nong, S.; Han, X.; Xiang, Y.; Qian, Y.; Wei, Y.; Zhang, T.; Tian, K.; Shen, K.; Yang, J.; Ma, X. Metabolic reprogramming in cancer: Mechanisms and therapeutics. MedComm (2020) 2023, 4, e218. [Google Scholar] [CrossRef]
- Lue, H.W.; Podolak, J.; Kolahi, K.; Cheng, L.; Rao, S.; Garg, D.; Xue, C.H.; Rantala, J.K.; Tyner, J.W.; Thornburg, K.L.; et al. Metabolic reprogramming ensures cancer cell survival despite oncogenic signaling blockade. Genes Dev. 2017, 31, 2067–2084. [Google Scholar] [CrossRef]
- Yang, J.; Shay, C.; Saba, N.F.; Teng, Y. Cancer metabolism and carcinogenesis. Exp. Hematol. Oncol. 2024, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The cancer metabolic reprogramming and immune response. Mol. Cancer 2021, 20, 28. [Google Scholar] [CrossRef] [PubMed]
- Broadfield, L.A.; Pane, A.A.; Talebi, A.; Swinnen, J.V.; Fendt, S.M. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev. Cell 2021, 56, 1363–1393. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Martin-Perez, M.; Urdiroz-Urricelqui, U.; Bigas, C.; Benitah, S.A. The role of lipids in cancer progression and metastasis. Cell Metab. 2022, 34, 1675–1699. [Google Scholar] [CrossRef] [PubMed]
- Hopperton, K.E.; Duncan, R.E.; Bazinet, R.P.; Archer, M.C. Fatty acid synthase plays a role in cancer metabolism beyond providing fatty acids for phospholipid synthesis or sustaining elevations in glycolytic activity. Exp. Cell Res. 2014, 320, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the wheels of the cancer machine: The role of lipid metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yan, Q.; Liu, X.; Wu, J. Unraveling lipid metabolism reprogramming for overcoming drug resistance in melanoma. Biochem. Pharmacol. 2024, 223, 116122. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, A.; Pilkington, S.M.; Rhodes, L.E. Ultraviolet-radiation induced skin inflammation: Dissecting the role of bioactive lipids. Chem. Phys. Lipids 2011, 164, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Nowowiejska, J.; Baran, A.; Flisiak, I. Lipid alterations and metabolism disturbances in selected inflammatory skin diseases. Int. J. Mol. Sci. 2023, 24, 7053. [Google Scholar] [CrossRef]
- Vergani, E.; Beretta, G.L.; Aloisi, M.; Costantino, M.; Corno, C.; Frigerio, S.; Tinelli, S.; Dugo, M.; Accattatis, F.M.; Granata, A.; et al. Targeting of the lipid metabolism impairs resistance to BRAF kinase inhibitor in melanoma. Front. Cell Dev. Biol. 2022, 10, 927118. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, L.; Albanesi, C.; Madonna, S. Recent updates on the involvement of PI3K/AKT/mTOR molecular cascade in the pathogenesis of hyperproliferative skin disorders. Front. Med. 2021, 8, 665647. [Google Scholar] [CrossRef] [PubMed]
- Sinnberg, T.; Lasithiotakis, K.; Niessner, H.; Schittek, B.; Flaherty, K.T.; Kulms, D.; Maczey, E.; Campos, M.; Gogel, J.; Garbe, C.; et al. Inhibition of PI3K-AKT-mTOR signaling sensitizes melanoma cells to cisplatin and temozolomide. J. Investig. Dermatol. 2009, 129, 1500–1515. [Google Scholar] [CrossRef]
- Wu, X.; Yu, J.; Yan, J.; Dai, J.; Si, L.; Chi, Z.; Sheng, X.; Cui, C.; Ma, M.; Tang, H.; et al. PI3K/AKT/mTOR pathway inhibitors inhibit the growth of melanoma cells with mTOR H2189Y mutations in vitro. Cancer Biol. Ther. 2018, 19, 584–589. [Google Scholar] [CrossRef]
- Dei Cas, M.; Ciniselli, C.M.; Vergani, E.; Ciusani, E.; Aloisi, M.; Duroni, V.; Verderio, P.; Ghidoni, R.; Paroni, R.; Perego, P.; et al. Alterations in plasma lipid profiles associated with melanoma and therapy resistance. Int. J. Mol. Sci. 2024, 25, 7053. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, L.; Morelli, M.; Scarponi, C.; Scaglione, G.L.; Pallotta, S.; Albanesi, C.; Madonna, S. PI3Kdelta sustains keratinocyte hyperproliferation and epithelial inflammation: Implications for a topically druggable target in psoriasis. Cells 2021, 10, 2336. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Schmelzle, T.; Hall, M.N. TOR, a central controller of cell growth. Cell 2000, 103, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Si, L.; Li, Y.; Wu, X.; Xu, X.; Dai, J.; Tang, H.; Ma, M.; Chi, Z.; Sheng, X.; et al. Analysis of mTOR gene aberrations in melanoma patients and evaluation of their sensitivity to PI3K-AKT-mTOR pathway inhibitors. Clin. Cancer Res. 2016, 22, 1018–1027. [Google Scholar] [CrossRef]
- Jebali, A.; Dumaz, N. The role of RICTOR downstream of receptor tyrosine kinase in cancers. Mol. Cancer 2018, 17, 39. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Willenborg, S.; Bloch, W.; Wickstrom, S.A.; Wagle, P.; Brodesser, S.; Roers, A.; Jais, A.; Bruning, J.C.; Hall, M.N.; et al. Epidermal mammalian target of rapamycin complex 2 controls lipid synthesis and filaggrin processing in epidermal barrier formation. J. Allergy Clin. Immunol. 2020, 145, 283–300.e288. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Y.; Wang, Y.; Zhang, W.; Wang, N.; Bai, R.; Luo, R.; Tuo, H.; Zheng, Y. LPCAT1 promotes melanoma cell proliferation via Akt signaling. Oncol. Rep. 2024, 51, 67. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Bok, I.; Jasani, N.; Wang, K.; Chadourne, M.; Mecozzi, N.; Deng, O.; Welsh, E.A.; Kinose, F.; Rix, U.; et al. PTEN lipid phosphatase activity suppresses melanoma formation by opposing an AKT/mTOR/FRA1 signaling axis. Cancer Res. 2024, 84, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Kuhajda, F.P. Fatty-acid synthase and human cancer: New perspectives on its role in tumor biology. Nutrition 2000, 16, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Li, T.; Zhang, X.; Gao, P.; Qiao, P.; Li, S.; Geng, Z. Expression and roles of fatty acid synthase in hepatocellular carcinoma. Oncol. Rep. 2014, 32, 2471–2476. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, H.; Li, J.; Fang, X.; Pan, H.; Yuan, X.; Zhang, P. Up-regulated FASN expression promotes transcoelomic metastasis of ovarian cancer cell through epithelial-mesenchymal transition. Int. J. Mol. Sci. 2014, 15, 11539–11554. [Google Scholar] [CrossRef]
- Wang, H.; Xi, Q.; Wu, G. Fatty acid synthase regulates invasion and metastasis of colorectal cancer via Wnt signaling pathway. Cancer Med. 2016, 5, 1599–1606. [Google Scholar] [CrossRef]
- Innocenzi, D.; Alo, P.L.; Balzani, A.; Sebastiani, V.; Silipo, V.; La Torre, G.; Ricciardi, G.; Bosman, C.; Calvieri, S. Fatty acid synthase expression in melanoma. J. Cutan. Pathol. 2003, 30, 23–28. [Google Scholar] [CrossRef]
- Talebi, A.; de Laat, V.; Spotbeen, X.; Dehairs, J.; Rambow, F.; Rogiers, A.; Vanderhoydonc, F.; Rizotto, L.; Planque, M.; Doglioni, G.; et al. Pharmacological induction of membrane lipid poly-unsaturation sensitizes melanoma to ROS inducers and overcomes acquired resistance to targeted therapy. J. Exp. Clin. Cancer Res. 2023, 42, 92. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, D.; Vallacchi, V.; Campi, V.; Ranzani, T.; Daniotti, M.; Chiodini, E.; Fiorentini, S.; Greeve, I.; Prinetti, A.; Rivoltini, L.; et al. DHCR24 gene expression is upregulated in melanoma metastases and associated to resistance to oxidative stress-induced apoptosis. Int. J. Cancer 2005, 115, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Pritchard-Jones, R.O.; Dunn, D.B.; Qiu, Y.; Varey, A.H.; Orlando, A.; Rigby, H.; Harper, S.J.; Bates, D.O. Expression of VEGF(xxx)b, the inhibitory isoforms of VEGF, in malignant melanoma. Br. J. Cancer 2007, 97, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Seguin, F.; Carvalho, M.A.; Bastos, D.C.; Agostini, M.; Zecchin, K.G.; Alvarez-Flores, M.P.; Chudzinski-Tavassi, A.M.; Coletta, R.D.; Graner, E. The fatty acid synthase inhibitor orlistat reduces experimental metastases and angiogenesis in B16-F10 melanomas. Br. J. Cancer 2012, 107, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Almeida, L.Y.; Bastos, D.C.; Ortega, R.M.; Moreira, F.S.; Seguin, F.; Zecchin, K.G.; Raposo, H.F.; Oliveira, H.C.; Amoedo, N.D.; et al. The fatty acid synthase inhibitor orlistat reduces the growth and metastasis of orthotopic tongue oral squamous cell carcinomas. Mol. Cancer Ther. 2014, 13, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Pandey, V.; Vijayakumar, M.V.; Ajay, A.K.; Malvi, P.; Bhat, M.K. Diet-induced obesity increases melanoma progression: Involvement of Cav-1 and FASN. Int. J. Cancer 2012, 130, 497–508. [Google Scholar] [CrossRef]
- Simiczyjew, A.; Wadzynska, J.; Pietraszek-Gremplewicz, K.; Kot, M.; Zietek, M.; Matkowski, R.; Nowak, D. Melanoma cells induce dedifferentiation and metabolic changes in adipocytes present in the tumor niche. Cell. Mol. Biol. Lett. 2023, 28, 58. [Google Scholar] [CrossRef]
- Tian, Y.; Ma, J.; Wang, M.; Yi, X.; Guo, S.; Wang, H.; Zhang, H.; Wang, H.; Yang, Y.; Zhang, B.; et al. BCKDHA contributes to melanoma progression by promoting the expressions of lipogenic enzymes FASN and ACLY. Exp. Dermatol. 2023, 32, 1633–1643. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Furukawa, K.; Hamamura, K.; Furukawa, K. Positive feedback loop between PI3K-Akt-mTORC1 signaling and the lipogenic pathway boosts Akt signaling: Induction of the lipogenic pathway by a melanoma antigen. Cancer Res. 2011, 71, 4989–4997. [Google Scholar] [CrossRef]
- Giampietri, C.; Petrungaro, S.; Cordella, M.; Tabolacci, C.; Tomaipitinca, L.; Facchiano, A.; Eramo, A.; Filippini, A.; Facchiano, F.; Ziparo, E. Lipid Storage and autophagy in melanoma cancer cells. Int. J. Mol. Sci. 2017, 18, 1271. [Google Scholar] [CrossRef] [PubMed]
- Dinavahi, S.S.; Chen, Y.C.; Gowda, R.; Dhanyamraju, P.K.; Punnath, K.; Desai, D.; Berg, A.; Kimball, S.R.; Amin, S.; Yang, J.M.; et al. Targeting protein translation in melanoma by inhibiting EEF-2 kinase regulates cholesterol metabolism though SREBP2 to inhibit tumour development. Int. J. Mol. Sci. 2022, 23, 3481. [Google Scholar] [CrossRef] [PubMed]
- Hamamura, K.; Furukawa, K.; Hayashi, T.; Hattori, T.; Nakano, J.; Nakashima, H.; Okuda, T.; Mizutani, H.; Hattori, H.; Ueda, M.; et al. Ganglioside GD3 promotes cell growth and invasion through p130Cas and paxillin in malignant melanoma cells. Proc. Natl. Acad. Sci. USA 2005, 102, 11041–11046. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, Y.; Miyazaki, S.; Hamamura, K.; Kambe, M.; Miyata, M.; Tajima, O.; Ohmi, Y.; Yamauchi, Y.; Furukawa, K.; Furukawa, K. Ganglioside GD3 enhances adhesion signals and augments malignant properties of melanoma cells by recruiting integrins to glycolipid-enriched microdomains. J. Biol. Chem. 2010, 285, 27213–27223. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Melloul, D.; Stoffel, M. Regulation of transcriptional coactivator PGC-1alpha. Sci. Aging Knowledge. Environ. 2004, 2004, pe9. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Ou, L.; Twaddel, W.; Fang, H.B.; Vafai, S.B.; Vazquez, F.; Puigserver, P.; Boros, L.; et al. PGC1alpha promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res. 2011, 71, 6888–6898. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Monks, B.; Ge, Q.; Birnbaum, M.J. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature 2007, 447, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- McGuirk, S.; Gravel, S.P.; Deblois, G.; Papadopoli, D.J.; Faubert, B.; Wegner, A.; Hiller, K.; Avizonis, D.; Akavia, U.D.; Jones, R.G.; et al. PGC-1alpha supports glutamine metabolism in breast cancer. Cancer Metab. 2013, 1, 22. [Google Scholar] [CrossRef]
- Cheng, C.F.; Ku, H.C.; Lin, H. PGC-1alpha as a pivotal factor in lipid and metabolic regulation. Int. J. Mol. Sci. 2018, 19, 3447. [Google Scholar] [CrossRef]
- Luo, C.; Lim, J.H.; Lee, Y.; Granter, S.R.; Thomas, A.; Vazquez, F.; Widlund, H.R.; Puigserver, P. A PGC1alpha-mediated transcriptional axis suppresses melanoma metastasis. Nature 2016, 537, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Yu, D.; Luo, C.; Bennett, C.; Jedrychowski, M.; Gygi, S.P.; Widlund, H.R.; Puigserver, P. Epigenetic suppression of PGC1alpha (PPARGC1A) causes collateral sensitivity to HMGCR-inhibitors within BRAF-treatment resistant melanomas. Nat. Commun. 2023, 14, 3251. [Google Scholar] [CrossRef] [PubMed]
- Gopal, Y.N.; Rizos, H.; Chen, G.; Deng, W.; Frederick, D.T.; Cooper, Z.A.; Scolyer, R.A.; Pupo, G.; Komurov, K.; Sehgal, V.; et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1alpha and oxidative phosphorylation in melanoma. Cancer Res. 2014, 74, 7037–7047. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef]
- Kridel, S.J.; Axelrod, F.; Rozenkrantz, N.; Smith, J.W. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004, 64, 2070–2075. [Google Scholar] [CrossRef]
- Carvalho, M.A.; Zecchin, K.G.; Seguin, F.; Bastos, D.C.; Agostini, M.; Rangel, A.L.; Veiga, S.S.; Raposo, H.F.; Oliveira, H.C.; Loda, M.; et al. Fatty acid synthase inhibition with Orlistat promotes apoptosis and reduces cell growth and lymph node metastasis in a mouse melanoma model. Int. J. Cancer 2008, 123, 2557–2565. [Google Scholar] [CrossRef]
- Ho, T.S.; Ho, Y.P.; Wong, W.Y.; Chi-Ming Chiu, L.; Wong, Y.S.; Eng-Choon Ooi, V. Fatty acid synthase inhibitors cerulenin and C75 retard growth and induce caspase-dependent apoptosis in human melanoma A-375 cells. Biomed. Pharmacother. 2007, 61, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Zecchin, K.G.; Rossato, F.A.; Raposo, H.F.; Melo, D.R.; Alberici, L.C.; Oliveira, H.C.; Castilho, R.F.; Coletta, R.D.; Vercesi, A.E.; Graner, E. Inhibition of fatty acid synthase in melanoma cells activates the intrinsic pathway of apoptosis. Lab Investig. 2011, 91, 232–240. [Google Scholar] [CrossRef]
- Bastos, D.C.; Paupert, J.; Maillard, C.; Seguin, F.; Carvalho, M.A.; Agostini, M.; Coletta, R.D.; Noel, A.; Graner, E. Effects of fatty acid synthase inhibitors on lymphatic vessels: An in vitro and in vivo study in a melanoma model. Lab Investig. 2017, 97, 194–206. [Google Scholar] [CrossRef]
- de Almeida, L.Y.; Mariano, F.S.; Bastos, D.C.; Cavassani, K.A.; Raphelson, J.; Mariano, V.S.; Agostini, M.; Moreira, F.S.; Coletta, R.D.; Mattos-Graner, R.O.; et al. The antimetastatic activity of orlistat is accompanied by an antitumoral immune response in mouse melanoma. Cancer Chemother. Pharmacol. 2020, 85, 321–330. [Google Scholar] [CrossRef]
- Stamatakos, S.; Beretta, G.L.; Vergani, E.; Dugo, M.; Corno, C.; Corna, E.; Tinelli, S.; Frigerio, S.; Ciusani, E.; Rodolfo, M.; et al. Deregulated FASN expression in BRAF inhibitor-resistant melanoma cells unveils new targets for drug combinations. Cancers 2021, 13, 2284. [Google Scholar] [CrossRef] [PubMed]
- Ventura, R.; Mordec, K.; Waszczuk, J.; Wang, Z.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G.; Heuer, T.S. Inhibition of de novo palmitate synthesis by fatty acid synthase induces apoptosis in tumor cells by remodeling cell membranes, inhibiting signaling pathways, and reprogramming gene expression. EBioMedicine 2015, 2, 808–824. [Google Scholar] [CrossRef] [PubMed]
- Heuer, T.S.; Ventura, R.; Mordec, K.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G. FASN Inhibition and taxane treatment combine to enhance anti-tumor efficacy in diverse xenograft tumor models through disruption of tubulin palmitoylation and microtubule organization and FASN inhibition-mediated effects on oncogenic signaling and gene expression. EBioMedicine 2017, 16, 51–62. [Google Scholar] [PubMed]
- Kamisuki, S.; Mao, Q.; Abu-Elheiga, L.; Gu, Z.; Kugimiya, A.; Kwon, Y.; Shinohara, T.; Kawazoe, Y.; Sato, S.; Asakura, K.; et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem. Biol. 2009, 16, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Talebi, A.; Dehairs, J.; Rambow, F.; Rogiers, A.; Nittner, D.; Derua, R.; Vanderhoydonc, F.; Duarte, J.A.G.; Bosisio, F.; Van den Eynde, K.; et al. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat. Commun. 2018, 9, 2500. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Shi, Y.; Feng, Z.; Yuan, D.; Guo, S.; Wang, Y.; Shen, H.; Li, Y.; Yan, F.; Wang, Y. Fatostatin promotes anti-tumor immunity by reducing SREBP2 mediated cholesterol metabolism in tumor-infiltrating T lymphocytes. Eur. J. Pharmacol. 2024, 971, 176519. [Google Scholar] [CrossRef]
- Tang, J.J.; Li, J.G.; Qi, W.; Qiu, W.W.; Li, P.S.; Li, B.L.; Song, B.L. Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab. 2011, 13, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Pisha, E.; Chai, H.; Lee, I.S.; Chagwedera, T.E.; Farnsworth, N.R.; Cordell, G.A.; Beecher, C.W.; Fong, H.H.; Kinghorn, A.D.; Brown, D.M.; et al. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nat. Med. 1995, 1, 1046–1051. [Google Scholar] [CrossRef]
- Tan, Y.; Yu, R.; Pezzuto, J.M. Betulinic acid-induced programmed cell death in human melanoma cells involves mitogen-activated protein kinase activation. Clin. Cancer Res. 2003, 9, 2866–2875. [Google Scholar]
- Gheorgheosu, D.; Jung, M.; Oren, B.; Schmid, T.; Dehelean, C.; Muntean, D.; Brune, B. Betulinic acid suppresses NGAL-induced epithelial-to-mesenchymal transition in melanoma. Biol. Chem. 2013, 394, 773–781. [Google Scholar] [CrossRef]
- Oliveira-Costa, J.F.; Meira, C.S.; Neves, M.; Dos Reis, B.; Soares, M.B.P. Anti-inflammatory activities of betulinic Acid: A review. Front. Pharmacol. 2022, 13, 883857. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Plonka, P.M.; Reiss, K. Melanoma-Time to fast or time to feast? An interplay between PPARs, metabolism and immunity. Exp. Dermatol. 2020, 29, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta. 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Zoico, E.; Darra, E.; Rizzatti, V.; Tebon, M.; Franceschetti, G.; Mazzali, G.; Rossi, A.P.; Fantin, F.; Zamboni, M. Role of adipose tissue in melanoma cancer microenvironment and progression. Int. J. Obes. 2018, 42, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.H.; Um, J.Y.; Lee, S.G.; Yang, W.M.; Sethi, G.; Ahn, K.S. Conditioned media from adipocytes promote proliferation, migration, and invasion in melanoma and colorectal cancer cells. J. Cell. Physiol. 2019, 234, 18249–18261. [Google Scholar] [CrossRef] [PubMed]
- Coelho, P.; Almeida, J.; Prudencio, C.; Fernandes, R.; Soares, R. Effect of adipocyte secretome in melanoma progression and vasculogenic mimicry. J. Cell Biochem. 2016, 117, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Wadzynska, J.; Simiczyjew, A.; Pietraszek-Gremplewicz, K.; Kot, M.; Zietek, M.; Matkowski, R.; Nowak, D. The impact of cellular elements of TME on melanoma biology and its sensitivity to EGFR and MET targeted therapy. Biochim. Biophys. Acta Mol. Cell Res. 2023, 1870, 119549. [Google Scholar] [CrossRef] [PubMed]
- Kwan, H.Y.; Fu, X.; Liu, B.; Chao, X.; Chan, C.L.; Cao, H.; Su, T.; Tse, A.K.W.; Fong, W.F.; Yu, Z.L. Subcutaneous adipocytes promote melanoma cell growth by activating the Akt signaling pathway: Role of palmitic acid. J. Biol. Chem. 2014, 289, 30525–30537. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Faouzi, S.; Souquere, S.; Roy, S.; Routier, E.; Libenciuc, C.; Andre, F.; Pierron, G.; Scoazec, J.Y.; Robert, C. Melanoma persister cells are tolerant to BRAF/MEK inhibitors via ACOX1-mediated fatty acid oxidation. Cell Rep. 2020, 33, 108421. [Google Scholar] [CrossRef]
- Wang, X.D.; Kim, C.; Zhang, Y.; Rindhe, S.; Cobb, M.H.; Yu, Y. Cholesterol regulates the tumor adaptive resistance to MAPK pathway inhibition. J. Proteome Res. 2021, 20, 5379–5391. [Google Scholar] [CrossRef]
- Fedida-Metula, S.; Elhyany, S.; Tsory, S.; Segal, S.; Hershfinkel, M.; Sekler, I.; Fishman, D. Targeting lipid rafts inhibits protein kinase B by disrupting calcium homeostasis and attenuates malignant properties of melanoma cells. Carcinogenesis 2008, 29, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Roh, W.; Sullivan, R.J.; Wong, K.H.K.; Wittner, B.S.; Guo, H.; Dubash, T.D.; Sade-Feldman, M.; Wesley, B.; Horwitz, E.; et al. The lipogenic regulator SREBP2 induces transferrin in circulating melanoma cells and suppresses ferroptosis. Cancer Discov. 2021, 11, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Falletta, P.; Goding, C.R.; Vivas-Garcia, Y. Connecting metabolic rewiring with phenotype switching in melanoma. Front. Cell Dev. Biol. 2022, 10, 930250. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Carrie, L.; Dufau, C.; Nieto, L.; Segui, B.; Levade, T.; Riond, J.; Andrieu-Abadie, N. Lipid metabolic reprogramming: Role in melanoma progression and therapeutic perspectives. Cancers 2020, 12, 3147. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, Y.; Tang, K.; Li, Z.; Halberstam, A.A.; Zhou, L.; Perry, R.J. Thiazolidinedione enhances the efficacy of anti-PD-1 monoclonal antibody in murine melanoma. Am. J. Physiol. Endocrinol. Metab. 2024, 326, E341–E350. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kook, E.; Kim, D.-H. Elucidating the Role of Lipid-Metabolism-Related Signal Transduction and Inhibitors in Skin Cancer. Metabolites 2024, 14, 309. https://doi.org/10.3390/metabo14060309
Kook E, Kim D-H. Elucidating the Role of Lipid-Metabolism-Related Signal Transduction and Inhibitors in Skin Cancer. Metabolites. 2024; 14(6):309. https://doi.org/10.3390/metabo14060309
Chicago/Turabian StyleKook, Eunjin, and Do-Hee Kim. 2024. "Elucidating the Role of Lipid-Metabolism-Related Signal Transduction and Inhibitors in Skin Cancer" Metabolites 14, no. 6: 309. https://doi.org/10.3390/metabo14060309
APA StyleKook, E., & Kim, D.-H. (2024). Elucidating the Role of Lipid-Metabolism-Related Signal Transduction and Inhibitors in Skin Cancer. Metabolites, 14(6), 309. https://doi.org/10.3390/metabo14060309