
Accurate Prediction of 1H NMR Chemical Shifts of Small Molecules Using Machine Learning


 and
and 
Abstract
1. Introduction
2. Methods
2.1. Creating a Solvent-Aware 1H Chemical Shift Dataset for Training and Validation
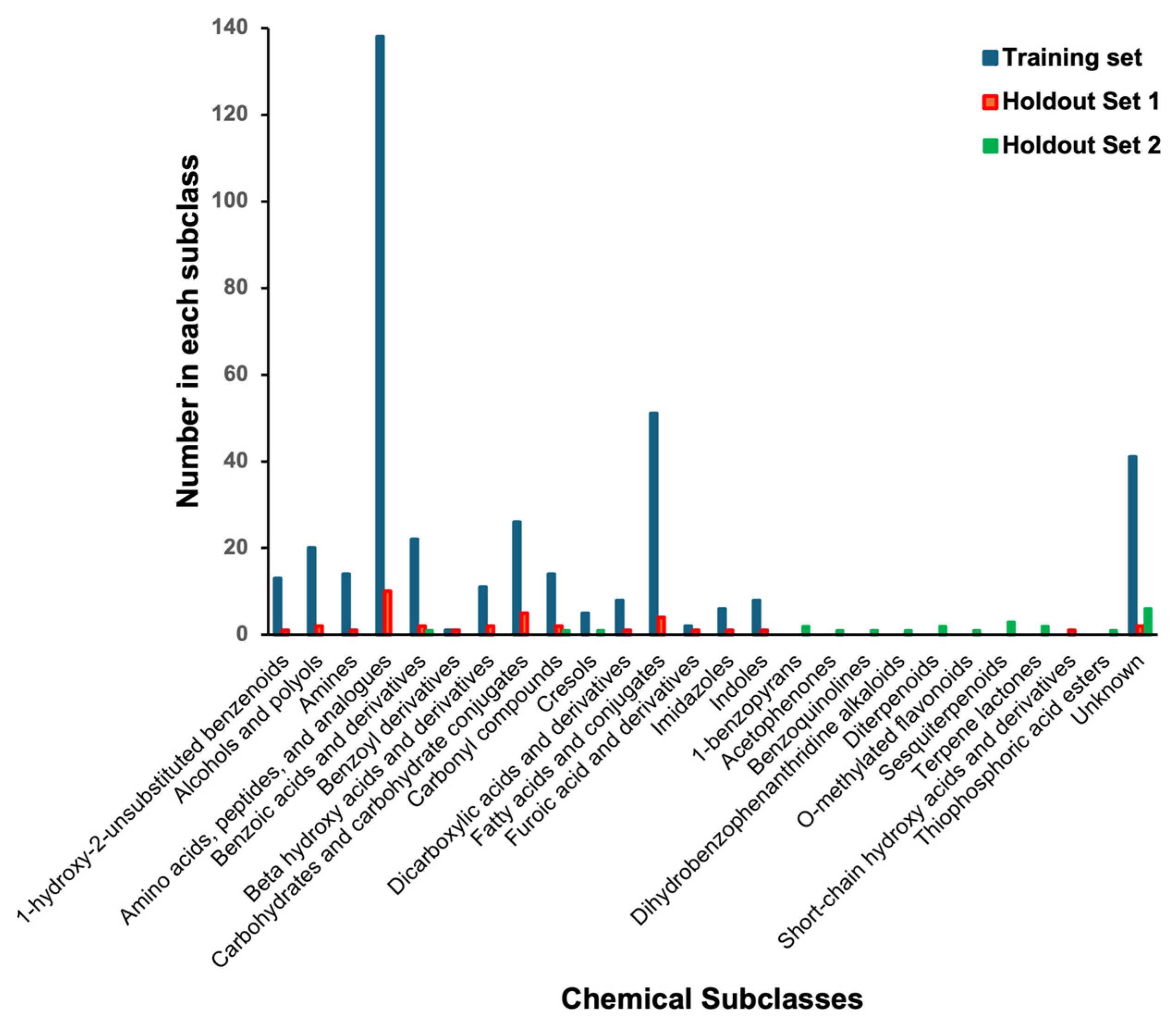
2.1.1. The Training Dataset
2.1.2. The Holdout Datasets
2.2. Data Cleaning and Correction
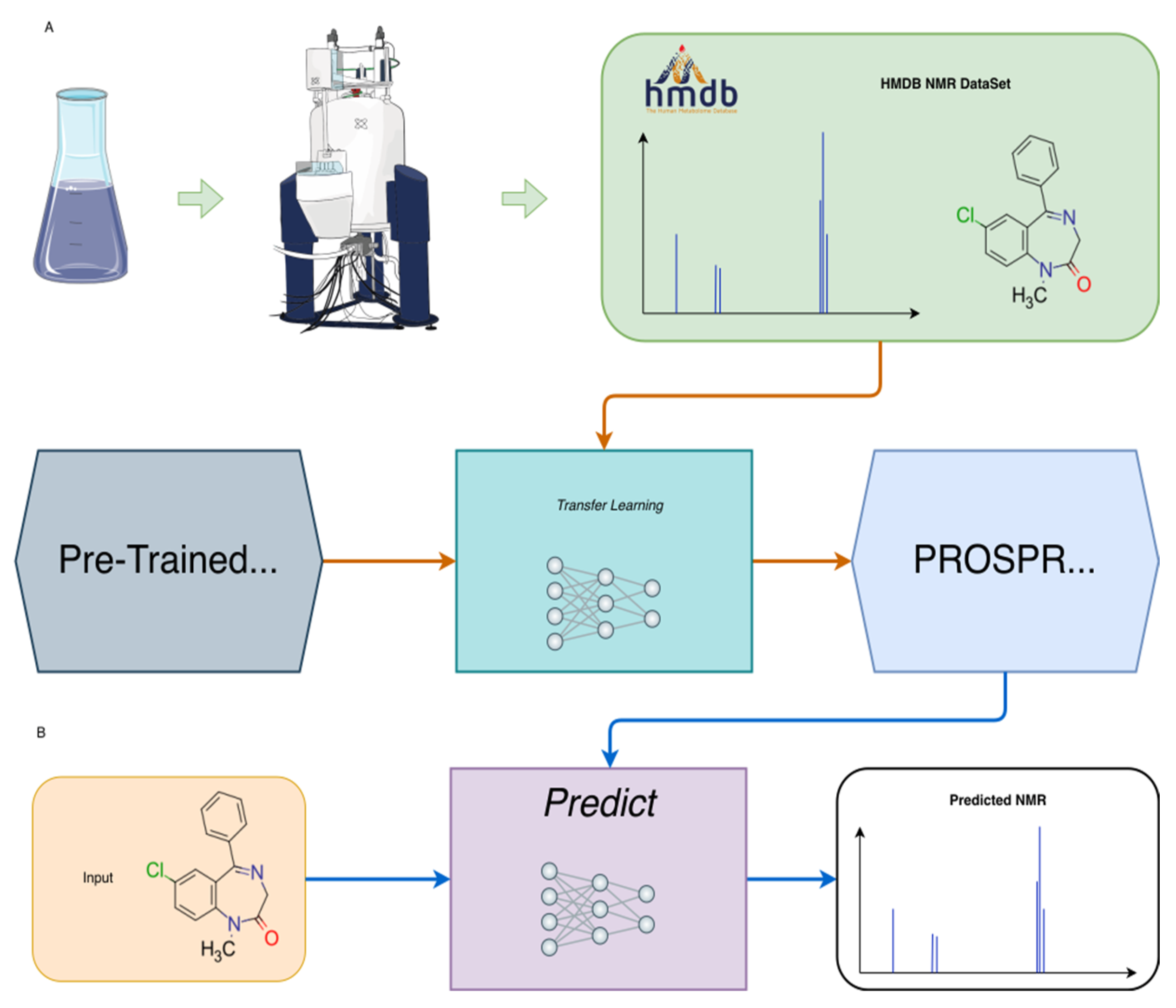
2.3. Machine Learning Method
2.4. 1H NMR Chemical Shift Predictions for Different Solvents and Internal Standards
3. Results
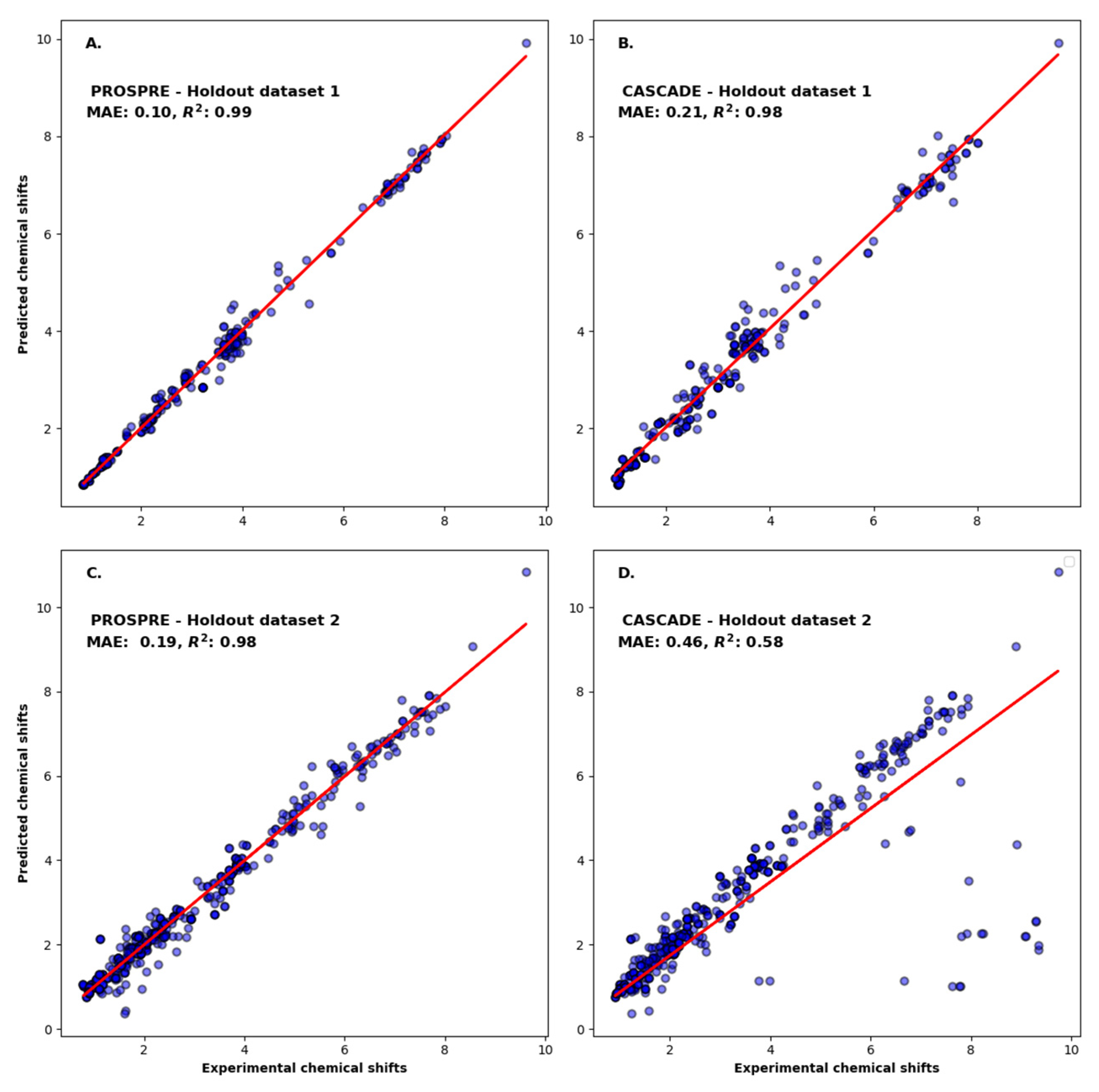
3.1. Performance Evaluation
3.2. Applications
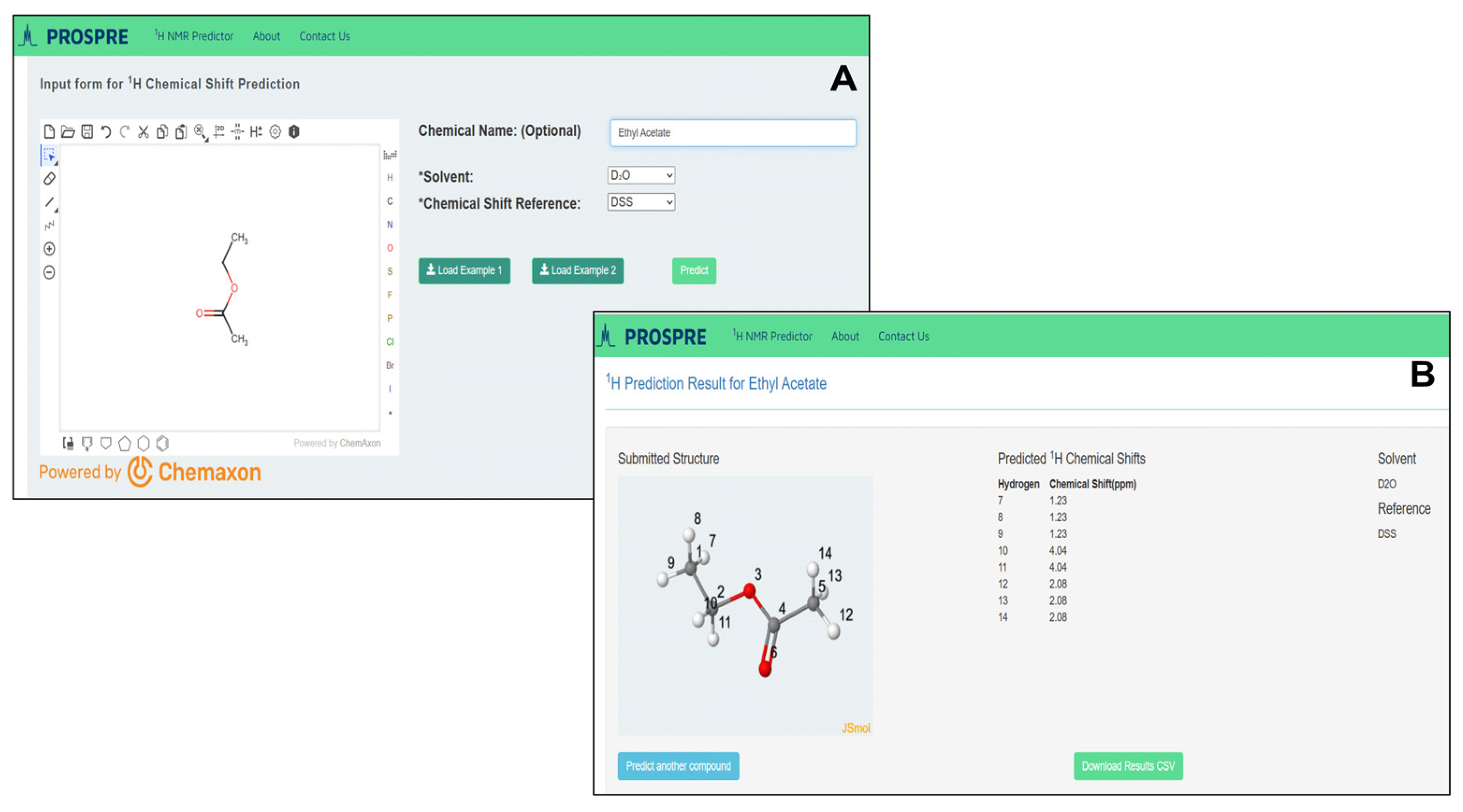
3.3. The PROSPRE Webserver
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anaraki, M.T.; Lysak, D.H.; Downey, K.; Kock, F.V.C.; You, X.; Majumdar, R.D.; Barison, A.; Lião, L.M.; Ferreira, A.G.; Decker, V.; et al. NMR spectroscopy of wastewater: A review, case study, and future potential. Prog. Nucl. Magn. Reson. Spectrosc. 2021, 126–127, 121–180. [Google Scholar] [CrossRef] [PubMed]
- Labine, L.M.; Simpson, M.J. The use of nuclear magnetic resonance (NMR) and mass spectrometry (MS)–based metabolomics in environmental exposure assessment. Curr. Opin. Environ. Sci. Health 2020, 15, 7–15. [Google Scholar] [CrossRef]
- Simpson, M.J.; Bearden, D.W. Environmental Metabolomics: NMR Techniques. In eMagRes; Harris, R.K., Wasylishen, R.L., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2013; Volume 2, pp. 549–560. ISBN 9780470034590. [Google Scholar]
- Shi, L.; Zhang, N. Applications of solution NMR in drug discovery. Molecules 2021, 26, 576. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, C.; Conde, R.; Embade, N.; Mato, J.M.; Millet, O. Metabolomics as a powerful tool for diagnostic, prognostic and drug intervention analysis in COVID-19. Front. Mol. Biosci. 2023, 10, 1111482. [Google Scholar] [CrossRef] [PubMed]
- Egan, J.M.; van Santen, J.A.; Liu, D.Y.; Linington, R.G. Development of an NMR-based platform for the direct structural annotation of complex natural products mixtures. J. Nat. Prod. 2021, 84, 1044–1055. [Google Scholar] [CrossRef]
- Wild, C.P.; Scalbert, A.; Herceg, Z. Measuring the exposome: A powerful basis for evaluating environmental exposures and cancer risk. Environ. Mol. Mutagen. 2013, 54, 480–499. [Google Scholar] [CrossRef] [PubMed]
- Wojtowicz, W.; Zabek, A.; Deja, S.; Dawiskiba, T.; Pawelka, D.; Glod, M.; Balcerzak, W.; Mlynarz, P. Serum and urine (1)H NMR-based metabolomics in the diagnosis of selected thyroid diseases. Sci. Rep. 2017, 7, 9108. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.R.; Kobayashi, N.; Wedell, J.R.; Baskaran, K.; Iwata, T.; Yokochi, M.; Maziuk, D.; Yao, H.; Fujiwara, T.; Kurusu, G.; et al. BioMagResBank (BMRB) as a resource for structural biology. Methods Mol. Biol. 2020, 2112, 187–218. [Google Scholar] [CrossRef]
- Steinbeck, C.; Krause, S.; Kuhn, S. NMRShiftDB—Constructing a free chemical information system with open-source components. J. Chem. Inf. Comput. Sci. 2003, 43, 1733–1739. [Google Scholar] [CrossRef]
- Saito, T.; Kinugasa, S. Development and release of a spectral database for organic compounds—Key to the continual services and success of a large-scale database. Synthesiology 2011, 4, 35–44. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sayeeda, Z.; Budinski, Z.; Guo, A.C.; Lee, B.L.; Berjanskii, M.; Rout, M.; Peters, H.; Dizon, R.; Mah, R.; et al. NP-MRD: The Natural Products Magnetic Resonance Database. Nucleic Acids Res. 2022, 50, D665–D677. [Google Scholar] [CrossRef]
- Knox, C.; Wilson, M.; Klinger, C.M.; Franklin, M.; Oler, E.; Wilson, A.; Pon, A.; Cox, J.; Chin, N.E.L.; Strawbridge, S.A.; et al. DrugBank 6.0: The DrugBank knowledgebase for 2024. Nucleic Acids Res. 2023, 52, D1265–D1275. [Google Scholar] [CrossRef]
- Lokhov, P.G.; Maslov, D.L.; Kharibin, O.N.; Balashova, E.E.; Archakov, A.I. Label-free data standardization for clinical metabolomics. BioData Min. 2017, 10, 10. [Google Scholar] [CrossRef]
- Nuñez, J.R.; Colby, S.M.; Thomas, D.G.; Tfaily, M.M.; Tolic, N.; Ulrich, E.M.; Sobus, J.R.; Metz, T.O.; Teeguarden, J.G.; Renslow, R.S. Advancing standards-free methods for the identification of small molecules in complex samples. arXiv 2018, arXiv:1810.07367. [Google Scholar] [CrossRef]
- Jonas, E.; Kuhn, S.; Schlörer, N. Prediction of chemical shift in NMR: A review. Magn. Reson. Chem. 2021, 60, 1021–1031. [Google Scholar] [CrossRef]
- Shoolery, J.N.; Rogers, M.T. Nuclear magnetic resonance spectra of steroids. J. Am. Chem. Soc. 1958, 80, 5121–5135. [Google Scholar] [CrossRef]
- Dailey, B.P.; Shoolery, J.N. The electron withdrawal power of substituent groups. J. Am. Chem. Soc. 1955, 77, 3977–3981. [Google Scholar] [CrossRef]
- Kalchhauser, H.; Robien, W. CSEARCH: A computer program for identification of organic compounds and fully automated assignment of carbon-13 nuclear magnetic resonance spectra. J. Chem. Inf. Comput. Sci. 1985, 25, 103. [Google Scholar] [CrossRef]
- Bremser, W. Hose—A novel substructure code. Anal. Chim. Acta 1978, 103, 355–365. [Google Scholar] [CrossRef]
- Kuhn, S.; Johnson, S.R. Stereo-aware extension of HOSE codes. ACS Omega 2019, 4, 7323–7329. [Google Scholar] [CrossRef]
- Bühl, M.; Kaupp, M.; Malkina, O.L.; Malkin, V.G. The DFT route to NMR chemical shifts. J. Comput. Chem. 1999, 20, 91–105. [Google Scholar] [CrossRef]
- Guan, Y.; Shree Sowndarya, S.V.; Gallegos, L.C.; St John, P.C.; Paton, R.S. Real-time prediction of 1H and 13C chemical shifts with DFT accuracy using a 3D graph neural network. Chem. Sci. 2021, 12, 12012–12026. [Google Scholar] [CrossRef] [PubMed]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef]
- Kvasnicka, V.; Sklenak, S.; Pospichal, J. Application of recurrent neural networks in chemistry. Prediction and classification of carbon-13 NMR chemical shifts in a series of monosubstituted benzenes. J. Chem. Inf. Comput. Sci. 1992, 32, 742–747. [Google Scholar] [CrossRef]
- Meiler, J.; Meusinger, R.; Will, M. Fast determination of 13C NMR chemical shifts using artificial neural networks. J. Chem. Inf. Comput. Sci. 2000, 40, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Aires-de-Sousa, J.; Hemmer, M.C.; Gasteiger, J. Prediction of 1H NMR chemical shifts using neural networks. Anal. Chem. 2002, 74, 80–90. [Google Scholar] [CrossRef]
- Binev, Y.; Aires-de-Sousa, J. Structure-based predictions of 1H NMR chemical shifts using feed-forward neural networks. J. Chem. Inf. Comput. Sci. 2004, 44, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Jonas, E.; Kuhn, S. Rapid prediction of NMR spectral properties with quantified uncertainty. J. Cheminform. 2019, 11, 50. [Google Scholar] [CrossRef]
- Schaefer, T.; Schneider, W.G. On the nature of solvent effects in the proton resonance spectra of unsaturated ring compounds. I. Substituted benzenes. J. Chem. Phys. 1960, 32, 1218–1222. [Google Scholar] [CrossRef]
- Matsuo, T. Studies of the solvent effect on the chemical shifts in n.m.r. spectroscopy. II. Solutions of succinic anhydride, maleic anhydride, and the N-substituted imides. Can. J. Chem. 1967, 45, 1829–1835. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Bigam, C.G.; Yao, J.; Abildgaard, F.; Dyson, H.J.; Oldfield, E.; Markley, J.L.; Sykes, B.D. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Dashti, H.; Wedell, J.R.; Westler, W.M.; Tonelli, M.; Aceti, D.; Amarasinghe, G.K.; Markley, J.L.; Eghbalnia, H.R. Applications of parametrized NMR spin systems of small molecules. Anal. Chem. 2018, 90, 10646–10649. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Sykes, B.D. Chemical shifts as a tool for structure determination. Methods Enzymol. 1994, 239, 363–392. [Google Scholar] [CrossRef] [PubMed]
- Dashti, H.; Westler, W.M.; Markley, J.L.; Eghbalnia, H.R. Unique identifiers for small molecules enable rigorous labeling of their atoms. Sci. Data 2017, 4, 170073. [Google Scholar] [CrossRef] [PubMed]
- Willcott, M.R. MestRe Nova. J. Am. Chem. Soc. 2009, 131, 13180. [Google Scholar] [CrossRef]
- Friebolin, H. Basic One-and Two-Dimensional NMR Spectroscopy, 4th ed.; Wiley-VCH: Weinheim, Germany, 2005; ISBN 3-527-31233-1. [Google Scholar]
- Rychnovsky, S.D. Predicting NMR spectra by computational methods: Structure revision of hexacyclinol. Org. Lett. 2006, 8, 2895–2898. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Soldi, C.; Jones, P.B.; Olmstead, M.M.; Rita, J.; Shaw, J.T.; Tantillo, D.J. The correct structure of aquatolide—Experimental validation of a theoretically-predicted structural revision. J. Am. Chem. Soc. 2012, 134, 18550–18553. [Google Scholar] [CrossRef]
- Hoffman, R. Magnetic susceptibility measurement by NMR: 2. The magnetic susceptibility of NMR solvents and their chemical shifts. J. Magn. Reson. 2022, 335, 107105. [Google Scholar] [CrossRef]
- Wishart, D.S.; Oler, E.; Peters, H.; Guo, A.; Girod, S.; Han, S.; Saha, S.; Lui, V.; LeVatte, M.; Gautam, V.; et al. MiMeDB: The Human Microbial Metabolome Database. Nucleic Acids Res. 2023, 51, D611–D620. [Google Scholar] [CrossRef] [PubMed]
- Sajed, T.; Marcu, A.; Ramirez, M.; Pon, A.; Guo, A.C.; Knox, C.; Wilson, M.; Grant, J.R.; Djoumbou, Y.; Wishart, D.S. ECMDB 2.0: A richer resource for understanding the biochemistry of E. coli. Nucleic Acids Res. 2016, 44, D495–D501. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Gaona, M.; Marcu, A.; Pon, A.; Guo, A.C.; Sajed, T.; Wishart, N.A.; Karu, N.; Feunang, Y.D.; Arndt, D.; Wishart, D.S. YMDB 2.0: A significantly expanded version of the yeast metabolome database. Nucleic Acids Res. 2017, 45, D440–D445. [Google Scholar] [CrossRef] [PubMed]
- Mohammed Taha, H.; Aalizadeh, R.; Alygizakis, N.; Antignac, J.P.; Arp, H.P.H.; Bade, R.; Baker, N.; Belova, L.; Bijlsma, L.; Bolton, E.E.; et al. The NORMAN Suspect List Exchange (NORMAN-SLE): Facilitating European and worldwide collaboration on suspect screening in high resolution mass spectrometry. Environ. Sci. Eur. 2022, 34, 104. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Pasin, D.; Skinnider, M.A.; Liigand, J.; Kleis, J.-N.; Brown, D.; Oler, E.; Sajed, T.; Gautam, V.; Harrison, S.; et al. Deep learning-enabled MS/MS spectrum prediction facilitates automated identification of novel psychoactive substances. Anal. Chem. 2023, 95, 18326–18334. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Brüschweiler, R. Knowns and unknowns in metabolomics identified by multidimensional NMR and hybrid MS/NMR methods. Curr. Opin. Biotechnol. 2017, 43, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Csizmadia, F. JChem: Java applets and modules supporting chemical database handling from web browsers. J. Chem. Inf. Comput. Sci. 2000, 40, 323–324. [Google Scholar] [CrossRef]
- Hanson, R.M. Jmol SMILES and Jmol SMARTS: Specifications and applications. J. Cheminform. 2016, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Herráez, A. Biomolecules in the computer: Jmol to the rescue. Biochem. Mol. Biol. Educ. 2006, 34, 255–261. [Google Scholar] [CrossRef]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform. 2016, 8, 61. [Google Scholar] [CrossRef]
- Wang, S.; Witek, J.; Landrum, G.A.; Riniker, S. Improving conformer generation for small rings and macrocycles based on distance geometry and experimental torsional-angle preferences. J. Chem. Inf. Model. 2020, 60, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method\Dataset | Holdout Dataset #1 (MAE) 1 | Holdout Dataset #2 (MAE) |
|---|---|---|
| PROSPRE | 0.10 ppm | 0.19 ppm |
| NMRShiftDB | 0.17 ppm | 0.25 ppm |
| MNOVA | 0.15 ppm | 0.20 ppm |
| CASCADE | 0.21 ppm | 0.46 ppm |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sajed, T.; Sayeeda, Z.; Lee, B.L.; Berjanskii, M.; Wang, F.; Gautam, V.; Wishart, D.S. Accurate Prediction of 1H NMR Chemical Shifts of Small Molecules Using Machine Learning. Metabolites 2024, 14, 290. https://doi.org/10.3390/metabo14050290
Sajed T, Sayeeda Z, Lee BL, Berjanskii M, Wang F, Gautam V, Wishart DS. Accurate Prediction of 1H NMR Chemical Shifts of Small Molecules Using Machine Learning. Metabolites. 2024; 14(5):290. https://doi.org/10.3390/metabo14050290
Chicago/Turabian StyleSajed, Tanvir, Zinat Sayeeda, Brian L. Lee, Mark Berjanskii, Fei Wang, Vasuk Gautam, and David S. Wishart. 2024. "Accurate Prediction of 1H NMR Chemical Shifts of Small Molecules Using Machine Learning" Metabolites 14, no. 5: 290. https://doi.org/10.3390/metabo14050290
APA StyleSajed, T., Sayeeda, Z., Lee, B. L., Berjanskii, M., Wang, F., Gautam, V., & Wishart, D. S. (2024). Accurate Prediction of 1H NMR Chemical Shifts of Small Molecules Using Machine Learning. Metabolites, 14(5), 290. https://doi.org/10.3390/metabo14050290