
Ketoacidosis and SGLT2 Inhibitors: A Narrative Review

 , ,
, ,  ,
, 

Abstract
1. Introduction
1.1. Diabetic Ketoacidosis
- Blood glucose levels >250 mg/dL;
- Arterial blood gases pH <7.3;
- Anion GAP >10 mEq/L;
- HCO3− <18 mEq/L;
- Ketones in blood and/or urine.
- Arterial pH <7.3;
- β-hydroxybutyrate ≥40 mg/dL (3.8 mmol/L) in adults;
- Ketone-positive urine (nitroprusside reaction method);
- Anion gap <10 mEq/L;
- Drowsy, stupor or coma in moderate to severe DKA [16].
- Hyperglycemia: blood glucose levels >11 mmol/L (200 mg/dL);
- Venous pH <7.3 or serum bicarbonate <15 mmol/L;
- Ketonemia and/or ketonuria.
- Mild: venous pH < 7.3 or serum bicarbonate <15 mmol/L;
- Moderate: pH < 7.2, serum bicarbonate <10 mmol/L;
- Severe: pH < 7.1, serum bicarbonate <5 mmol/L.
- Diabetic people with normal or not particularly raised glucose levels;
- The development of raised anion gap metabolic acidosis;
- Ketonemia (>3.0 mmol/L) or significant ketonuria (2+ or more on standard urine sticks) [15].
1.2. Sodium/Glucose Co-Transporter-2 Inhibitors
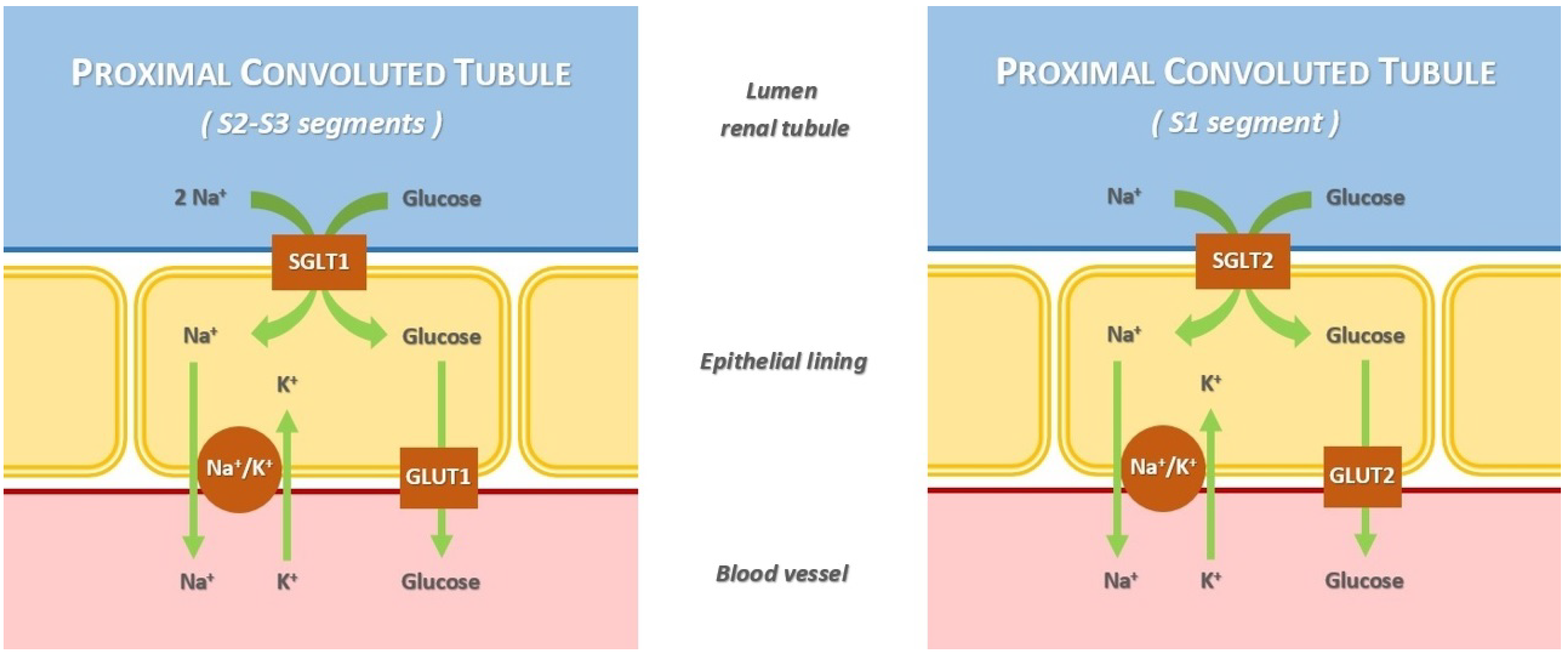
- SGLT type 1 (SGLT1) is a high-affinity low-capacity transporter, encoded by the SCL5A1 gene. It is located in the S2 and S3 segments of the renal proximal tubule. The percentage of renal glucose absorption by SGLT1 is almost 10%; the ratio of glucose and sodium co-transportation is 1:2. Its principal extrarenal locations are the gastrointestinal tract; in fact, the clinical syndrome resulting from SCL5A1 gene mutation has diarrhea as the dominant symptom. Heart and red blood cells are other extrarenal sites.
- SGLT type 2 (SGLT2) is a low-affinity high-capacity transporter, encoded by the SCL5A2 gene. It is present mainly in the S1 segment of the proximal tubule, near Bowman’s capsule. It reabsorbs almost 90% of the filtered glucose; the ratio of glucose and sodium co-transportation is 1:1. Its extrarenal locations are the brain and liver [20,21].
2. Epidemiology
3. Etiopathogenesis
Precipitating Factors
- Age of onset younger than 50 years;
- Acute symptoms (e.g., polyuria, polydipsia, unintentional weight loss);
- Body mass index less than 25 Kg/m2;
- A personal history of autoimmune disease;
- A family history of autoimmune disease.
4. Management
- A reduction in the blood ketone concentration by 0.5 mmol/L/h;
- An increase in venous bicarbonate by 3.0 mmol/L/h;
- Lowering capillary blood glucose by 3.0 mmol/L/h;
- Maintaining potassium between 4.0 and 5.5 mmol/L.
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2013, 36 (Suppl. S1), S67–S74. [Google Scholar] [CrossRef]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Aroda, V.R.; Collins, B.S.; Gabbay, R.A.; Green, J.; Maruthur, N.M.; Rosas, S.E.; Del Prato, S.; Mathieu, C.; Mingrone, G.; et al. Management of hyperglycaemia in type 2 diabetes, 2022. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2022, 65, 1925–1966. [Google Scholar] [CrossRef]
- Cole, J.B.; Florez, J.C. Genetics of diabetes and diabetes complications. Nat. Rev. Nephrol. 2020, 16, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, B.K.; Srivastava, A.K. Diabetes mellitus: Complications and therapeutics. Med. Sci. Monit. 2006, 12, 130–147. [Google Scholar] [PubMed]
- Nyenwe, E.A.; Kitabchi, A.E. The evolution of diabetic ketoacidosis: An update of its etiology, pathogenesis and management. Metabolism 2016, 65, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Lapolla, A.; Amaro, F.; Bruttomesso, D.; Di Bartolo, P.; Grassi, G.; Maffeis, C.; Purrello, F.; Tumini, S. Diabetic Ketoacidosis: A Consensus Statement of the Associazione Medici Diabetologi (AMD), Societa Italiana di Diabetologia (SID), Societa Italiana di Endocrinologia e Diabetologia Pediatrica (SIEDP). Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1633–1644. [Google Scholar] [CrossRef] [PubMed]
- Kitabchi, A.E.; Umpierrez, G.E.; Miles, J.M.; Fisher, J.N. Hyperglycemic crises in adult patients with diabetes. Diabetes Care 2009, 32, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Westerberg, D.P. Diabetic ketoacidosis: Evaluation and treatment. Am. Fam. Physician 2013, 87, 337–346. [Google Scholar]
- Dhatariya, K.K.; Umpierrez, G.E. Guidelines for management of diabetic ketoacidosis: Time to revise? Lancet Diabetes Endocrinol. 2017, 5, 321–323. [Google Scholar] [CrossRef]
- Dhatariya, K.K.; Vellanki, P. Treatment of Diabetic Ketoacidosis (DKA)/Hyperglycemic Hyperosmolar State (HHS): Novel Advances in the Management of Hyperglycemic Crises (UK Versus USA). Curr. Diabetes Rep. 2017, 17, 33. [Google Scholar] [CrossRef]
- Misra, S.; Oliver, N.S. Diabetic ketoacidosis in adults. BMJ 2015, 351, h5660. [Google Scholar] [CrossRef]
- El Sayed, N.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Hilliard, M.E.; Isaacs, D.; Johnson, E.L.; et al. 2. Classification and Diagnosis of Diabetes: Standards of Care in Diabetes 2023. Diabetes Care 2023, 46 (Suppl. S1), S19–S40. [Google Scholar] [CrossRef]
- Savage, M.W.; Dhatariya, K.K.; Kilvert, A.; Rayman, G.; Rees, J.A.; Courtney, C.H.; Hilton, L.; Dyer, P.H.; Hamersley, M.S.; Joint British Diabetes Societies. Joint British Diabetes Societies guideline for the management of diabetic ketoacidosis. Diabet. Med. 2011, 28, 508–515. [Google Scholar] [CrossRef]
- Dhatariya, K.K.; Joint British Diabetes Societies for Inpatient Care. The management of diabetic ketoacidosis in adults-An updated guideline from the Joint British Diabetes Society for Inpatient Care. Diabet. Med. 2022, 39, e14788. [Google Scholar] [CrossRef]
- Handelsman, Y.; Henry, R.R.; Bloomgarden, Z.T.; Dagogo-Jack, S.; DeFronzo, R.A.; Einhorn, D.; Ferrannini, E.; Fonseca, V.A.; Garber, A.J.; Grunberger, G.; et al. American Association of Clinical Endocrinologists and American College of Endocrinology Position Statement On The Association Of SGLT-2 Inhibitors and Diabetic Ketoacidosis. Endocr. Pract. 2016, 22, 753–762. [Google Scholar] [CrossRef]
- Munro, J.F.; Campbell, I.W.; McCuish, A.C.; Duncan, L.J. Euglycaemic diabetic ketoacidosis. Br. Med. J. 1973, 2, 578–580. [Google Scholar] [CrossRef]
- Peters, A.L.; Buschur, E.O.; Buse, J.B.; Cohan, P.; Diner, J.C.; Hirsch, I.B. Euglycemic diabetic ketoacidosis: A potential complication of treatment with sodium-glucose cotransporter 2 inhibition. Diabetes Care 2015, 38, 1687–1693. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.A.; Norton, L.; Defronzo, R.A. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr. Rev. 2011, 32, 515–531. [Google Scholar] [CrossRef]
- Sarzani, R.; Giulietti, F.; Di Pentima, C.; Spannella, F. Sodium-glucose co-transporter-2 inhibitors: Peculiar “hybrid”diuretics that protect from target organ damage and cardiovascular events. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1622–1632. [Google Scholar] [CrossRef]
- Chao, E.C.; Henry, R.R. SGLT2 inhibition: A novel strategy for diabetes treatment. Nat. Rev. Drug Discov. 2010, 9, 551–559. [Google Scholar] [CrossRef]
- Jung, E.C.; Jang, J.E.; Park, J.Y. A Novel Therapeutic Agent for Type 2 Diabetes Mellitus: SGLT2 Inhibitor. Diabetes Metab. J. 2014, 38, 261–273. [Google Scholar] [CrossRef]
- Brown, E.; Rajeev, S.P.; Cuthbertson, D.J.; Wilding, J.P.H. A review of the mechanism of action, metabolic profile and haemodynamic effects of sodium-glucose co-transporter-2 inhibitors. Diabetes Obes. Metab. 2019, 21 (Suppl. S2), 9e18. [Google Scholar] [CrossRef]
- Rahmoune, H.; Thompson, P.W.; Ward, J.M.; Smith, C.D.; Hong, G.; Brown, J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 2005, 54, 3427–3434. [Google Scholar] [CrossRef]
- De Fronzo, R.A.; Davidson, J.A.; Del Prato, S. The role of the kidneys in glucose homeostasis: A new path towards normalizing glycaemia. Diabetes Obes. Metab. 2012, 14, 5–14. [Google Scholar] [CrossRef]
- Gronda, E.G.; Vanoli, E.; Iacoviello, M. Renal effects of SGLT2 inhibitors in cardiovascular patients with and without chronic kidney disease: Focus on heart failure and renal outcomes. Heart Fail. Rev. 2023, 28, 723–732. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and renal outcomes in Type 2 diabetes and nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Zannad, F.; Ferreira, J.P.; Pocock, S.J.; Anker, S.D.; Butler, J.; Filippatos, G.; Brueckmann, M.; Ofstad, A.P.; Pfarr, E.; Jamal, W.; et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: A meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet 2020, 396, 819–829. [Google Scholar] [CrossRef]
- Gaborit, B.; Ancel, P.; Abdullah, A.E.; Maurice, F.; Abdesselam, I.; Calen, A.; Soghomonian, A.; Houssays, M.; Varlet, I.; Eisinger, M.; et al. Effect of empagliflozin on ectopic fat stores and myocardial energetics in type 2 diabetes: The EMPACEF study. Cardiovasc. Diabetol. 2021, 20, 57. [Google Scholar] [CrossRef] [PubMed]
- Filippatos, G.; Butler, J.; Farmakis, D.; Zannad, F.; Ofstad, A.P.; Ferreira, J.P.; Green, J.B.; Rosenstock, J.; Schnaidt, S.; Brueckmann, M.; et al. Empagliflozin for Heart Failure With Preserved Left Ventricular Ejection Fraction With and Without Diabetes. Circulation 2022, 146, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.; Di Bartolo, P.; Candido, R.; Lucisano, G.; Manicardi, V.; Giandalia, A.; Nicolucci, A.; Rocca, A.; Rossi, M.C.; Di Cianni, G.; et al. The AMD ANNALS: A continuous initiative for the improvement of type 2 diabetes care. Diabetes Res. Clin. Pract. 2023, 199, 110672. [Google Scholar] [CrossRef]
- Perkovic, V.; De Zeeuw, D.; Mahaffey, K.V.; Fulcher, G.; Erondu, N.; Shaw, W.; Barrett, T.D.; Weidner-Wells, M.; Deng, H.; Matthews, D.R.; et al. Canagliflozin and renal outcomes in type 2 diabetes: Results from the CANVAS Program randomised clinical trials. Lancet Diabetes Endocrinol. 2018, 6, 691–704. [Google Scholar] [CrossRef]
- Food and Drug Administration. Drug Safety Communication: FDA Revises Labels of SGLT2 Inhibitors for Diabetes to Include Warnings about too Much Acid in the Blood and Serious Urinary Tract Infections. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-revises-labels-sglt2-inhibitors-diabetes-include-warnings-about-too-much-acid-blood-and-serious (accessed on 16 March 2022).
- Sampani, E.; Sarafidis, P.; Papagianni, A. Euglycaemic diabetic ketoacidosis as a complication of SGLT-2 inhibitors: Epidemiology, pathophysiology, and treatment. Expert Opin. Drug Saf. 2020, 19, 673–682. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. EMA Confirms Recommendations to Minimise Ketoacidosis Risk with SGLT2 Inhibitors for Diabetes. Available online: https://www.ema.europa.eu/en/documents/referral/sglt2-inhibitors-article-20-procedure-ema-confirms-recommendations-minimise-ketoacidosis-risk-sglt2_en.pdf (accessed on 28 April 2016).
- Modi, A.; Agrawal, A.; Morgan, F. Euglycemic Diabetic Ketoacidosis: A Review. Curr. Diabetes Rev. 2017, 13, 315–321. [Google Scholar] [CrossRef]
- The EMPA-KIDNEY Collaborative Group; Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; et al. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 117–127. [Google Scholar]
- Peikert, A.; Martinez, F.A.; Vaduganathan, M.; Claggett, B.L.; Kulac, I.J.; Desai, A.S.; Jhund, P.S.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; et al. Efficacy and Safety of Dapagliflozin in Heart Failure With Mildly Reduced or Preserved Ejection Fraction According to Age: The DELIVER Trial. Circ. Heart Fail. 2022, 15, e010080. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Erondu, N.; Desai, M.; Ways, K.; Meininger, G. Diabetic ketoacidosis and related events in the canagliflozin type 2 diabetes clinical program. Diabetes Care 2015, 38, 1680–1686. [Google Scholar] [CrossRef]
- Cannon, C.P.; Pratley, R.; Dagogo-Jack, S.; Mancuso, J.; Huyck, S.; Masiukiewicz, U.; Charbonnel, B.; Frederich, R.; Gallo, S.; Cosentino, F.; et al. Cardiovascular Outcomes with Ertugliflozin in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 1425–1435. [Google Scholar] [CrossRef]
- Blau, J.E.; Tella, S.H.; Taylor, S.I.; Rother, K.I. Ketoacidosis associated with SGLT-2 inhibitor treatment: Analysis of FAERS data. Diabetes Metab. Res. Rev. 2017, 33, e2924. [Google Scholar] [CrossRef]
- Dhatariya, K.K. Defining and characterising diabetic ketoacidosis in adults. Diabetes Res. Clin. Pract. 2019, 155, 107797. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA Strengthens Kidney Warnings for Diabetes Medicines Canagliflozin (Invokana, In-vokamet) and Dapagliflozin (Farxiga, Xigduo XR). Available online: https://www.fda.gov/downloads/Drugs/DrugSafety/UCM506772.pdf (accessed on 17 June 2016).
- Fadini, G.P.; Bonora, B.M.; Avogaro, A. SGLT2 inhibitors and diabetic ketoacidosis: Data from the FDA adverse event reporting system. Diabetologia 2017, 60, 1385–1389. [Google Scholar] [CrossRef]
- He, Z.; Lam, K.; Zhao, W.; Yang, S.; Li, Y.; Mo, J.; Gao, S.; Liang, D.; Qiu, K.; Huang, M.; et al. SGLT-2 inhibitors and euglycemic diabetic ketoacidosis/diabetic ketoacidosis in FAERS: A pharmacovigilance assessment. Acta Diabetol. 2023, 60, 401–411. [Google Scholar] [CrossRef]
- Di Mauro, G.; Mascolo, A.; Gaio, M.; Rafaniello, C.; De Angelis, A.; Berrino, L.; Paolisso, G.; Rossi, F.; Capuano, A. The Reporting Frequency of Ketoacidosis Events with Dapagliflozin from the European Spontaneous Reporting System: The DAPA-KETO Study. Pharmaceuticals 2022, 15, 286. [Google Scholar] [CrossRef]
- Douros, A.; Lix, L.M.; Fralick, M.; Dell’Aniello, S.; Shah, B.R.; Ronksley, P.E.; Tremblay, É.; Hu, N.; Alessi-Severini, S.; Fisher, A.; et al. Sodium-Glucose Cotransporter-2 Inhibitors and the Risk for Diabetic Ketoacidosis: A Multicenter Cohort Study. Ann. Intern. Med. 2020, 173, 417–425. [Google Scholar] [CrossRef]
- Ueda, P.; Svanström, H.; Melbye, M.; Eliasson, B.; Svensson, A.M.; Franzén, S.; Gudbjörnsdottir, S.; Hveem, K.; Jonasson, C.; Pasternak, B. Sodium glucose cotransporter 2 inhibitors and risk of serious adverse events: Nationwide register based cohort study. BMJ 2018, 363, k4365. [Google Scholar] [CrossRef]
- Fralick, M.; Schneeweiss, S.; Patorno, E. Risk of diabetic ketoacidosis after initiation of an SGLT2 inhibitor. N. Engl. J. Med. 2017, 376, 2300–2302. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, P.S.; Wong, R.; Ekinci, E.I.; Fourlanos, S.; Shah, S.; Jones, A.R.; Hare, M.J.L.; Calder, G.L.; Epa, D.S.; George, E.M.; et al. SGLT2 inhibitors increase the risk of diabetic ketoacidosis developing in the community and during hospital admission. J. Clin. Endocrinol. Metab. 2019, 104, 3077–3087. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Desai, M.; Ryan, P.B.; DeFalco, F.J.; Schuemie, M.J.; Stang, P.E.; Berlin, J.A.; Yuan, Z. Incidence of diabetic ketoacidosis among patients with type 2 diabetes mellitus treated with SGLT2 inhibitors and other antihyperglycemic agents. Diabetes Res. Clin. Pract. 2017, 128, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.L.; Persson, F.; Andersen, G.S.; Ridderstråle, M.; Nolan, J.J.; Carstensen, B.; Jørgensen, M.E. Incidence of ketoacidosis in the Danish type 2 diabetes population before and after introduction of sodium-glucose cotransporter 2 inhibitors-a nationwide, retrospective cohort study, 1995–2014. Diabetes Care 2017, 40, e57–e58. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Lee, K.W.; Kim, H.J. Sodium-glucose co-transporter-2 inhibitors and the risk of ketoacidosis in patients with type 2 diabetes mellitus: A nationwide population-based cohort study. Diabetes Obes. Metab. 2018, 20, 1852–1858. [Google Scholar] [CrossRef]
- Kohler, S.; Zeller, C.; Iliev, H.; Kaspers, S. Safety and Tolerability of Empagliflozin in Patients with Type 2 Diabetes: Pooled Analysis of Phase I-III Clinical Trials. Adv. Ther. 2017, 34, 1707–1726. [Google Scholar] [CrossRef] [PubMed]
- Rigato, M.; Fadini, G.P.; Avogaro, A. Safety of sodium-glucose cotransporter 2 inhibitors in elderly patients with type 2 diabetes: A meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2023, 25, 2963–2969. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Li, D.; Wang, T.; Zhai, S.; Song, Y. Effect of sodium-glucose cotransporter 2 inhibitors on diabetic ketoacidosis among patients with type 2 diabetes: A meta-analysis of randomized controlled trials. Diabetes Care 2016, 39, e123–e124. [Google Scholar] [CrossRef]
- Nakhleh, A.; Othman, A.; Masri, A.; Zloczower, M.; Zolotov, S.; Shehadeh, N. Clinical Outcomes of Diabetic Ketoacidosis in Type 2 Diabetes Patients with and without SGLT2 Inhibitor Treatment: A Retrospective Study. Biomedicines 2023, 11, 2689. [Google Scholar] [CrossRef]
- Umapathysivam, M.M.; Gunton, J.; Stranks, S.N.; Jesudason, D. Euglycemic Ketoacidosis in Two Patients Without Diabetes After Introduction of Sodium–Glucose Cotransporter 2 Inhibitor for Heart Failure With Reduced Ejection Fraction. Diabetes Care 2024, 47, 140–143. [Google Scholar] [CrossRef]
- Cartwright, M.M.; Hajja, W.; Al-Khatib, S.; Hazeghazam, M.; Sreedhar, D.; Li, R.N.; Wong-McKinstry, E.; Carlson, R.W. Toxigenic and metabolic causes of ketosis and ketoacidotic syndromes. Crit. Care Clin. 2012, 28, 601–631. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Ferrannini, E. Euglycemic diabetic ketoacidosis: A predictable, detectable, and preventable safety concern with SGLT2 inhibitors. Diabetes Care 2015, 38, 1638–1642. [Google Scholar] [CrossRef]
- Ogawa, W.; Sakaguchi, K. Euglycemic diabetic ketoacidosis induced by SGLT2 inhibitors: Possible mechanism and contributing factors. J. Diabetes Investig. 2016, 7, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Hine, J.; Paterson, H.; Abrol, E.; Russell-Jones, D.; Herring, R. SGLT inhibition and euglycaemic diabetic ketoacidosis. Lancet Diabetes Endocrinol. 2015, 3, 503–504. [Google Scholar] [CrossRef] [PubMed]
- Bonner, C.; Kerr-Conte, J.; Gmyr, V.; Queniat, G.; Moerman, E.; Thévenet, J.; Beaucamps, C.; Delalleau, N.; Popescu, I.; Malaisse, W.J.; et al. Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat. Med. 2015, 21, 512–517. [Google Scholar] [CrossRef]
- Kibbey, R.G. SGLT-2 inhibition and glucagon: Cause for alarm? Trends Endocrinol. Metab. 2015, 26, 337–338. [Google Scholar] [CrossRef] [PubMed]
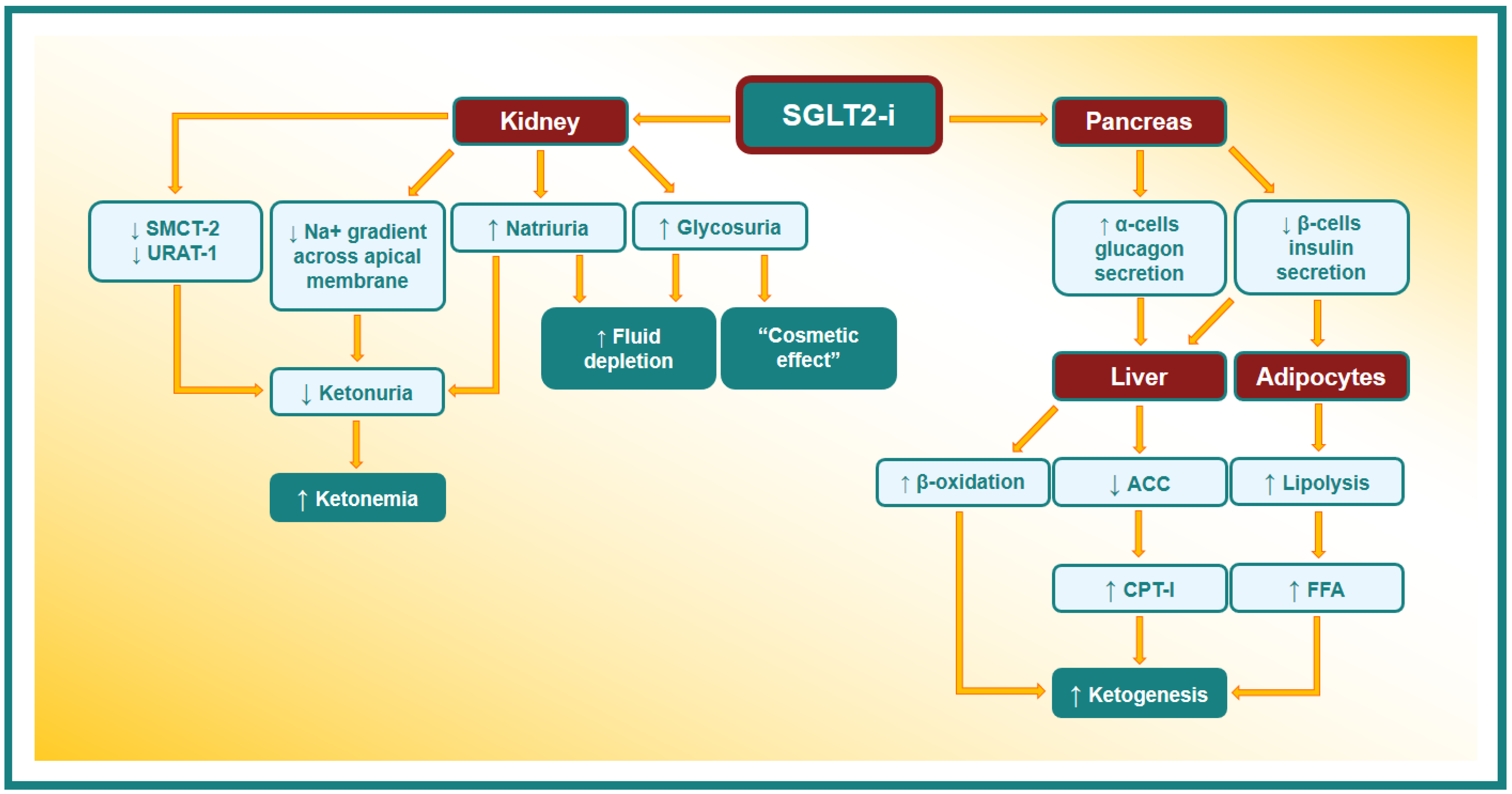
- Yu, X.; Zhang, S.; Zhang, L. Newer Perspectives of Mechanisms for Euglycemic Diabetic Ketoacidosis. Int. J. Endocrinol. 2018, 2018, 7074868. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M. Glucose transport families SLC5 and SLC50. Mol. Asp. Med. 2013, 34, 183–196. [Google Scholar] [CrossRef]
- Qiu, H.; Novikov, A.; Vallon, V. Ketosis and diabetic ketoacidosis in response to SGLT2 inhibitors: Basic mechanisms and therapeutic perspectives. Diabetes Metab. Res. Rev. 2017, 33, e2886. [Google Scholar] [CrossRef]
- Mandal, A.K.; Mount, D.B. The molecular physiology of uric acid homeostasis. Annu. Rev. Physiol. 2015, 77, 323–345. [Google Scholar] [CrossRef]
- Sarafidis, P.; Ferro, C.J.; Morales, E.; Ortiz, A.; Malyszko, J.; Hojs, R.; Khazim, K.; Ekart, R.; Valdivielso, J.; Fouque, D.; et al. SGLT-2 inhibitors and GLP-1 receptor agonists for nephroprotection and cardioprotection in patients with diabetes mellitus and chronic kidney disease. A consensus statement by the EURECA-m and the DIABESITY working groups of the ERA-EDTA. Nephrol. Dial. Transplant. 2019, 34, 208–230. [Google Scholar] [CrossRef]
- Pessoa, T.D.; Campos, L.C.; Carraro-Lacroix, L.; Girardi, A.C.; Malnic, G. Functional role of glucose metabolism, osmotic stress, and sodium-glucose cotransporter isoform-mediated transport on Na+/H+ exchanger isoform 3 activity in the renal proximal tubule. J. Am. Soc. Nephrol. 2014, 25, 2028–2039. [Google Scholar] [CrossRef]
- Rajeev, S.P.; Wilding, J. SGLT2 inhibition and ketoacidosis—Should we be concerned? Br. J. DiabetesVascular Dis. 2015, 15, 155–158. [Google Scholar] [CrossRef]
- D’Elia, J.A.; Segal, A.R.; Bayliss, G.P.; Weinrauch, L. Sodium-glucose cotransporter-2 inhibition and acidosis in patients with type 2 diabetes: A review of US FDA data and possible conclusions. Int. J. Nephrol. Renov. Dis. 2017, 10, 153–158. [Google Scholar] [CrossRef]
- Devenny, J.J.; Godonis, H.E.; Harvey, S.J.; Rooney, S.; Cullen, M.J.; Pelleymounter, M.A. Weight loss induced by chronic dapagliflozin treatment is attenuated by compensatory hyperphagia in diet-induced obese (DIO) rats. Obesity 2012, 20, 1645–1652. [Google Scholar] [CrossRef]
- Chow, E.; Clement, S.; Garg, R. Euglycemic diabetic ketoacidosis in the era of SGLT-2 inhibitors. BMJ Open Diabetes Res. Care 2023, 11, e003666. [Google Scholar] [CrossRef]
- Musso, G.; Saba, F.; Cassader, M.; Gambino, R. Diabetic ketoacidosis with SGLT2 inhibitors. BMJ 2020, 371, m4147. [Google Scholar] [CrossRef]
- Nasa, P.; Chaudhary, S.; Shrivastava, P.K.; Singh, A. Euglycemic diabetic ketoacidosis: A missed diagnosis. World J. Diabetes 2021, 12, 514–523. [Google Scholar] [CrossRef]
- Ata, F.; Yousaf, Z.; Khan, A.A.; Razok, A.; Akram, J.; Ali, E.A.H.; Abdalhadi, A.; Ibrahim, D.A.; Al Mohanadi, D.H.S.H.; Danjuma, M.I. SGLT-2 inhibitors associated euglycemic and hyperglycemic DKA in a multicentric cohort. Sci. Rep. 2021, 11, 10293. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Vellipuram, A.R.; Bandaru, S.S.; Pradeep Raj, J. Euglycemic diabetic ketoacidosis: A diagnostic and therapeutic dilemma. Endocrinol. Diabetes Metab. Case Rep. 2017, 2017, 17-0081. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F.; Clegg, D.J. Euglycemic Ketoacidosis as a Complication of SGLT2 Inhibitor Therapy. Clin. J. Am. Soc. Nephrol. 2021, 16, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Lentz, S.; Gottlieb, M. Alcoholic Ketoacidosis: Etiologies, Evaluation, and Management. J. Emerg. Med. 2021, 61, 658–665. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. SGLT2 Inhibitors: PRAC Makes Recommendations to Minimise Risk of Diabetic Ketoacidosis. Available online: https://www.ema.europa.eu/en/news/sglt2-inhibitors-prac-makes-recommendations-minimise-risk-diabetic-ketoacidosis (accessed on 12 February 2016).
- Goldenberg, R.M.; Berard, L.D.; Cheng, A.Y.Y.; Gilbert, J.D.; Verma, S.; Woo, V.C.; Yale, J.F. SGLT2 inhibitor-associated diabetic ketoacidosis: Clinical review and recommendations for prevention and diagnosis. Clin. Ther. 2016, 38, 2654–2664.e1. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, J.A.; Laugesen, E.; Leslie, R.D. Should there be concern about autoimmune diabetes in adults? Current evidence and controversies. Curr. Diabetes Rep. 2016, 16, 82. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.R.; Schumacher, C.A.; Spencer, E. HarpeSGLT2 Inhibitors: A Systematic Review of Diabetic Ketoacidosis and Related Risk Factors in the Primary Literature. Pharmacotherapy 2017, 37, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.S.; Epstein, S.; Corkey, B.E.; Grant, S.F.; Gavin, J.R.; Aguilar, R.B. The time is right for a new classification system for diabetes: Rationale and implications of the beta-cell-centric classification schema. Diabetes Care 2016, 39, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Rasouli, N.; Younes, N.; Ghosh, A.; Albu, J.; Cohen, R.M.; DeFronzo, R.A.; Diaz, E.; Sayyed Kassem, L.; Luchsinger, J.A.; McGill, J.B.; et al. Longitudinal effects of glucose-lowering medications on β-cell responses and insulin sensitivity in type 2 diabetes: The grade randomized clinical trial. Diabetes Care 2024, dc231070. [Google Scholar] [CrossRef] [PubMed]
- Sibai, B.M.; Viteri, O.A. Diabetic ketoacidosis in pregnancy. Obstet. Gynecol. 2014, 123, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Fourlanos, S.; Perry, C.; Stein, M.S.; Stankovich, J.; Harrison, L.C.; Colman, P.G. A clinical screening tool identifies autoimmune diabetes in adults. Diabetes Care 2006, 29, 970–975. [Google Scholar] [CrossRef]
- Bell, D.S.H.; Ovalle, F. The role of C-peptide levels in screening for latent autoimmune diabetes in adults. Am. J. Ther. 2004, 11, 308–311. [Google Scholar] [CrossRef]
- Karslioglu French, E.; Donihi, A.C.; Korytkowski, M.T. Diabetic ketoacidosis and hyperosmolar hyperglycemic syndrome: Review of acute decompensated diabetes in adult patients. BMJ 2019, 365, 1114. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.G.; Shrestha, E.; Wood, E.M. Euglycemic diabetic ketoacidosis: The paradox of delayed correction of acidosis. Diabetes Metab. Syndr 2023, 17, 102848. [Google Scholar] [CrossRef] [PubMed]
- Self, W.H.; Evans, C.S.; Jenkins, C.A.; Brown, R.M.; Casey, J.D.; Collins, S.P.; Coston, T.D.; Felbinger, M.; Flemmons, L.N.; Hellervik, S.M.; et al. Clinical Effects of Balanced Crystalloids vs Saline in Adults With Diabetic Ketoacidosis A Subgroup Analysis of Cluster Randomized Clinical Trials. JAMA Netw. Open 2020, 3, e2024596. [Google Scholar] [CrossRef] [PubMed]
- Wolfsdorf, J.I.; Glaser, N.; Agus, M.; Fritsch, M.; Hanas, R.; Rewers, A.; Sperling, M.A.; Codner, E. ISPAD Clinical Practice Consensus Guidelines 2018: Diabetic ketoacidosis and the hyperglycemic hyperosmolar state. Pediatr. Diabetes 2018, 19, 155–177. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, M.; Delaney, A.; Venkatesh, B. Fluid therapy in diabetic ketoacidosis. Curr. Opin. Clin. Nutr. Metab. Care 2024, 27, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Dhatariya, K.K.; Nunney, I.; Higgins, K.; Sampson, M.J.; Iceton, G. A national survey of the management of diabetic ketoacidosis in the UK in 2014. Diabet. Med. 2016, 33, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Evans, K. Diabetic ketoacidosis: Update on management. Clin. Med. 2019, 19, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Karajgikar, N.D.; Manroa, P.; Acharya, R.; Codario, R.A.; Reider, J.A.; Donihi, A.C.; Salata, R.A.; Korytkowski, M.T. Addressing pitfalls in management of diabetic ketoacidosis with a standardized protocol. Endocr. Pract. 2019, 25, 407–412. [Google Scholar]
- Duhon, B.; Attridge, R.L.; Franco-Martinez, A.C.; Maxwell, P.R.; Hughes, D.W. Intravenous sodium bicarbonate therapy in severely acidotic diabetic ketoacidosis. Ann. Pharmacother. 2013, 47, 970–975. [Google Scholar] [CrossRef]
- Liu, P.Y.; Jeng, C.Y. Severe hypophosphatemia in a patient with diabetic ketoacidosis and acute respiratory failure. JCMA 2013, 67, 355–359. [Google Scholar]
- Marini, H.R. Mediterranean Diet and Soy Isoflavones for Integrated Management of the Menopausal Metabolic Syndrome. Nutrients 2022, 14, 1550. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| JBDS Guidelines | ADA Guidelines | AACE/ACE Guidelines | AMD, SID and SIEDP Consensus Statement | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Mild | Moderate | Severe | Mild | Moderate | Severe | ||||
| “D” Glucose | >11.0 mmol/L (200 mg/dL) | >13.9 mmol/L (>250 mg/dL) | >13.9 mmol/L (>250 mg/dL) | >13.9 mmol/L (>250 mg/dL) | ___ | >11.0 mmol/L (200 mg/dL) | >11.0 mmol/L (200 mg/dL) | >11.0 mmol/L (200 mg/dL) | |
| “K” Ketones | >3.0 mmol/L (or history of diabetes) | Ketone-positive urine or serum | Ketone-positive urine or serum | Ketone-positive urine or serum | Ketone- positive serum ≥40 mg/dL (3.8 mmol/L) or urine | Ketonemia ≥3 mmol/L | Ketonemia ≥3 mmol/L | Ketonemia ≥3 mmol/L | |
| Ketonuria ≥2+ | Ketonuria ≥2+ | Ketonuria ≥2+ | |||||||
| “A” Acidosis | pH | <7.3 | 7.25–7.3 | 7.0–7.24 | <7.0 | <7.3 | <7.3 | <7.2 | <7.1 |
| HCO3 (mmol/L) | <15 | 15 to 18 | 10 to 15 | <10 | __ | <15 | <10 | <5 | |
| Anion gap (mEq/L) | ___ | >10 | >12 | >12 | >10 | ___ | ___ | ___ | |
| Mental Status | ___ | Alert | Alert or drowsy | Stupor or coma | Drowsy, stupor or coma | ___ | ___ | ___ | |
| Munro’s Classic Definition | JBDS Guidelines | |
|---|---|---|
| “D” Glucose | <16.6 mmol/L (300 mg/dL) | <13.8 mmol/L (250 mg/dL) |
| “K” Ketones | ___ | Ketonemia ≥31.6 mg/dL (3.0 mmol/L) Ketonuria (2+ or more on standard urine sticks) |
| “A” Acidosis | Raised anion gap metabolic acidosis | HCO3− <10 mEq/l |
| Medical History | ___ | Known diabetes history |
| TRIAL | SGLT2-i | Incidence in SGLT2-i Groups | Incidence in Placebo Groups | Hazard Ratio (95% CI) | p Values |
|---|---|---|---|---|---|
| EMPAREG-OUTCOME | Empagliflozin 10 mg Empagliflozin 25 mg | 1/2345 (<0.1%) 3/2342 (0.1%) | 1/2333 (<0.1%) | ___ | NS |
| EMPEROR-Preserved | Empagliflozin 10 mg | 4/1465 (0.3%) | 5/1471 (0.3%) | ___ | ___ |
| EMPA-KIDNEY | Empagliflozin 10 mg | 6/3304 (0.2%) 0.09/100 patients-yr | 1/3305 (<0.1%) 0.02/100 patients-yr | ___ | ___ |
| DECLARE-TIMI 58 | Dapagliflozin 10 mg | 27/8574 (0.3%) | 12/8569 (0.1%) | 2.18 (1.10–4.30) | 0.02 |
| DAPA-HF | Dapagliflozin 10 mg | 3/2373 * 0.1/100 patients-yr | 0/2371 | ___ | ___ |
| DELIVER | Dapagliflozin 10 mg | 0/3132 | 2/3131 (<0.1%) | ___ | ___ |
| DAPA-CKD | Dapagliflozin 10 mg | 0/2149 | 2/2149 (<0.1%) | ___ | 0.5 |
| CANVAS | Canagliflozin 100 mg Canagliflozin 300 mg | 0.6/1000 patients-yr | 0.3/1000 patients-yr | 2–33 (0.76–7.17) | 0.14 |
| CREDENCE | Canagliflozin 100 mg | 11/2200 2.2/1000 patients-yr | 1/2197 0.2/1000 patients-yr | 10.8 (1.39–83.65) | NA ** |
| VERTIS-CV | Ertugliflozin 5 mg Ertugliflozin 15 mg | 7/2746 (0.3) 12/2747 (0.4) | 2/2745 (0.1) | ___ | ___ |
| Precipitating Factors | Etiopathogenetic Mechanisms |
|---|---|
| Prolonged physical activity or exercise [70] |
|
| Decompensated cirrhosis [70,81] |
|
| Organic pancreatic insufficiency (acute or chronic pancreatitis) [70,81] |
|
| Fasting [70,83] |
|
| Acute cardio-vascular events (ACS or stroke) [81] |
|
| Cocaine use [81] |
|
| Trauma [81] |
|
| Infection, sepsis [81,83,84] |
|
| Excessive alcohol use [81,85] |
|
| Surgery [86,87] |
|
| LADA [88,89,90,91] |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morace, C.; Lorello, G.; Bellone, F.; Quartarone, C.; Ruggeri, D.; Giandalia, A.; Mandraffino, G.; Minutoli, L.; Squadrito, G.; Russo, G.T.; et al. Ketoacidosis and SGLT2 Inhibitors: A Narrative Review. Metabolites 2024, 14, 264. https://doi.org/10.3390/metabo14050264
Morace C, Lorello G, Bellone F, Quartarone C, Ruggeri D, Giandalia A, Mandraffino G, Minutoli L, Squadrito G, Russo GT, et al. Ketoacidosis and SGLT2 Inhibitors: A Narrative Review. Metabolites. 2024; 14(5):264. https://doi.org/10.3390/metabo14050264
Chicago/Turabian StyleMorace, Carmela, Giuseppe Lorello, Federica Bellone, Cristina Quartarone, Domenica Ruggeri, Annalisa Giandalia, Giuseppe Mandraffino, Letteria Minutoli, Giovanni Squadrito, Giuseppina T. Russo, and et al. 2024. "Ketoacidosis and SGLT2 Inhibitors: A Narrative Review" Metabolites 14, no. 5: 264. https://doi.org/10.3390/metabo14050264
APA StyleMorace, C., Lorello, G., Bellone, F., Quartarone, C., Ruggeri, D., Giandalia, A., Mandraffino, G., Minutoli, L., Squadrito, G., Russo, G. T., & Marini, H. R. (2024). Ketoacidosis and SGLT2 Inhibitors: A Narrative Review. Metabolites, 14(5), 264. https://doi.org/10.3390/metabo14050264