


Computational Applications: Beauvericin from a Mycotoxin into a Humanized Drug

 , ,
, ,  ,
,  , , and
, , and
Abstract
1. Beauvericin: A Mycotoxin
2. Arthropodpathogenic Activity of Beauvericin
3. From Mycotoxin to Humanized Drug
3.1. The Role of BEA in Antimicrobial Resistance
3.2. The Antiviral Activity of BEA
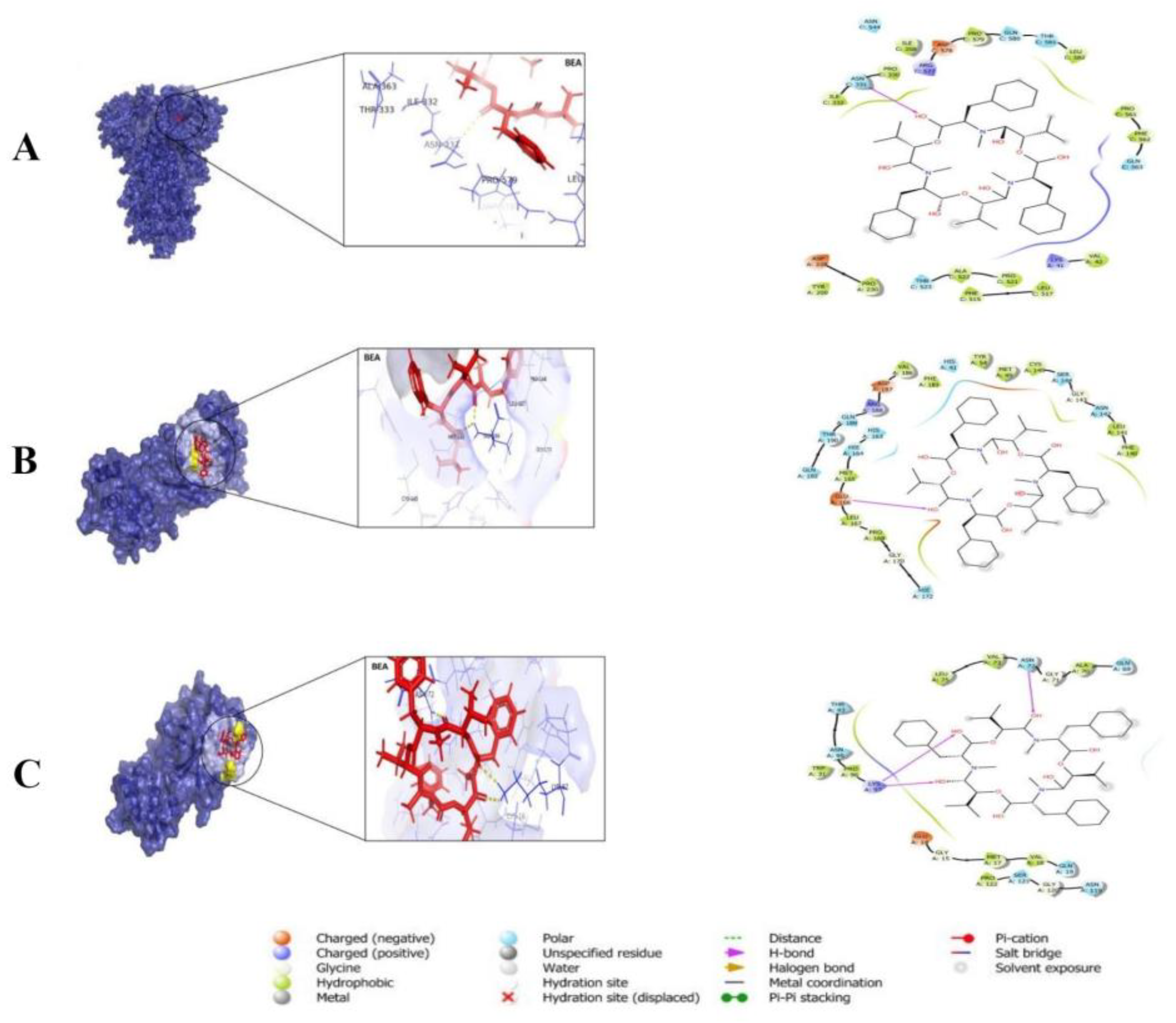
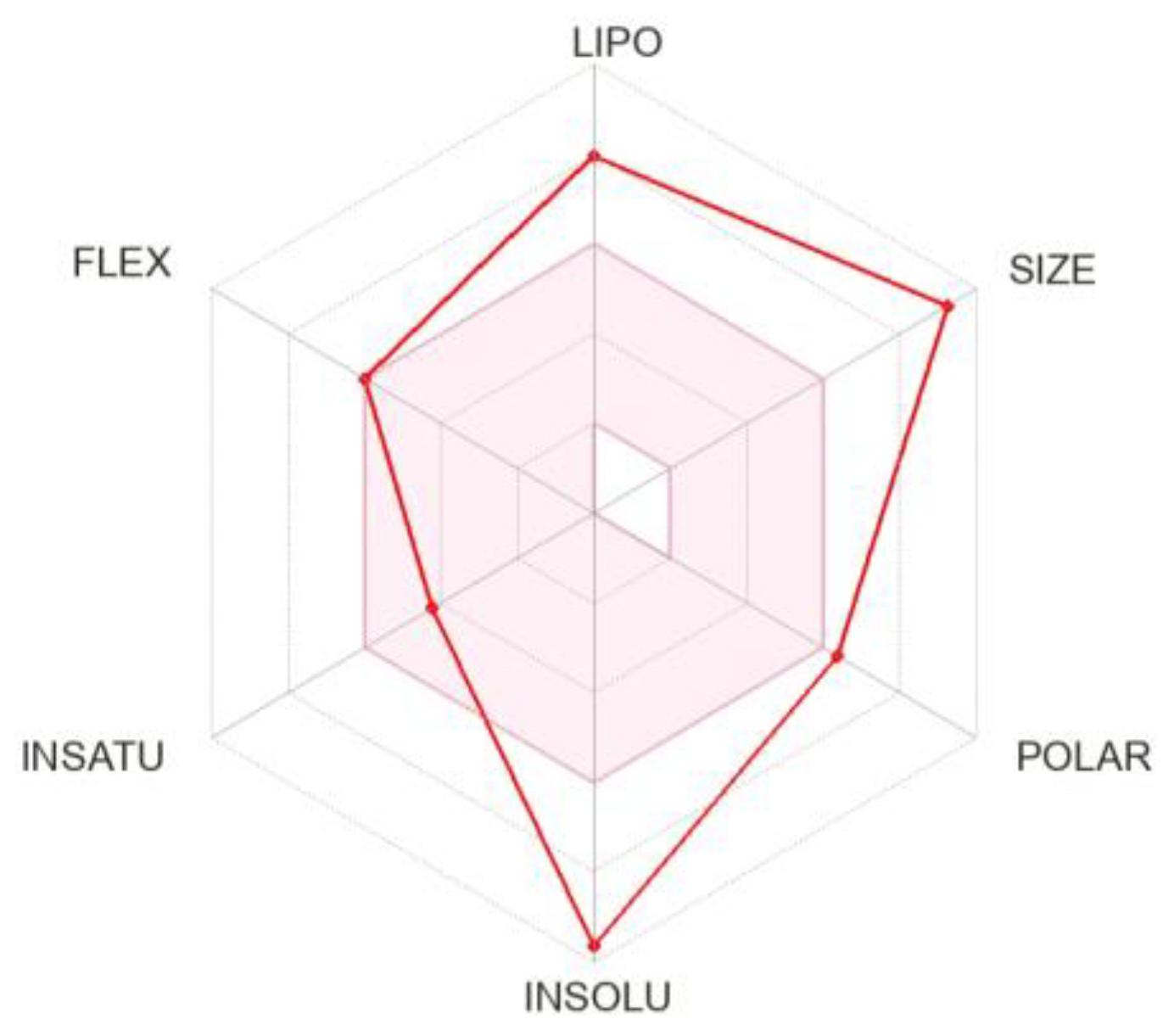
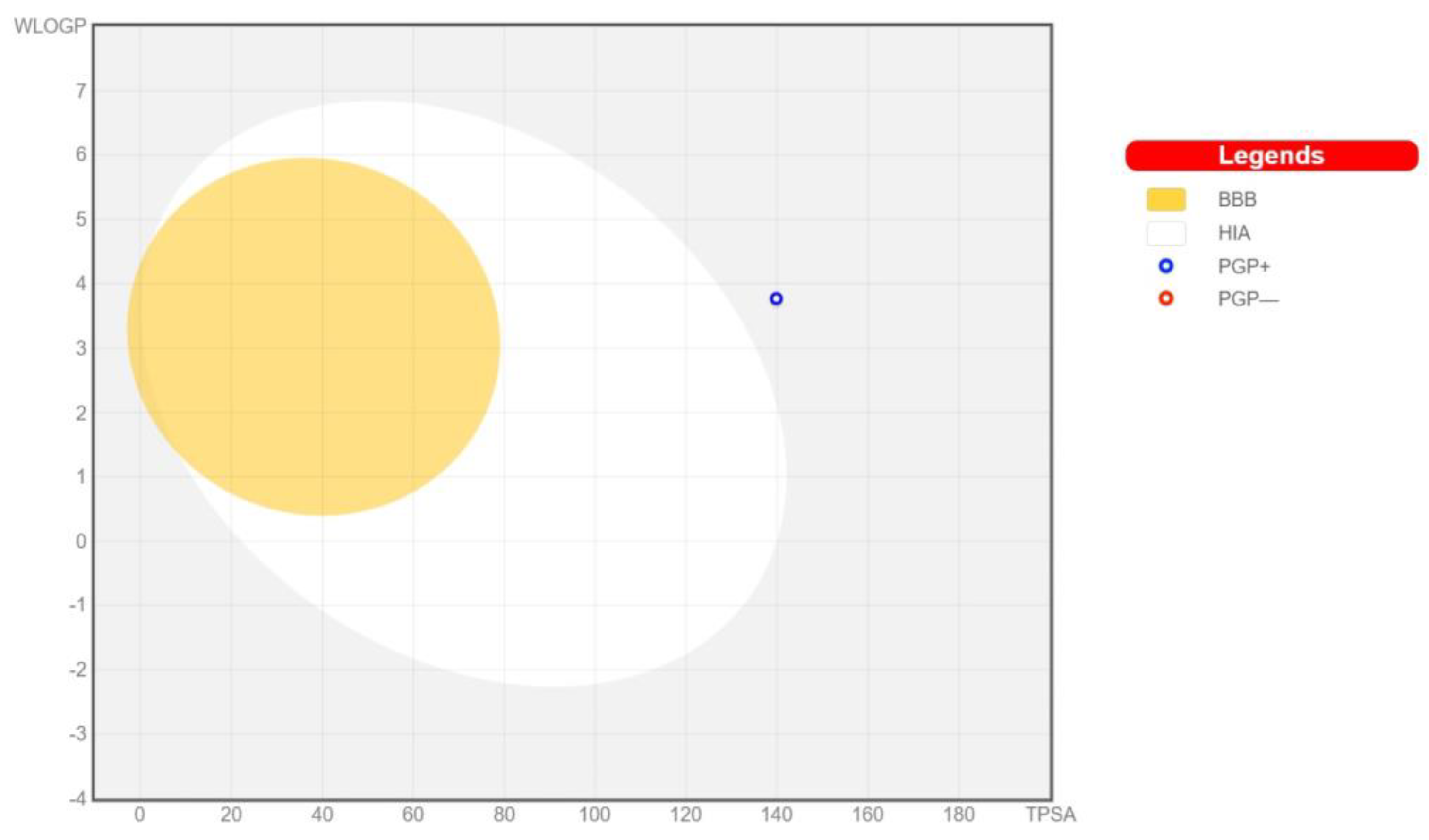
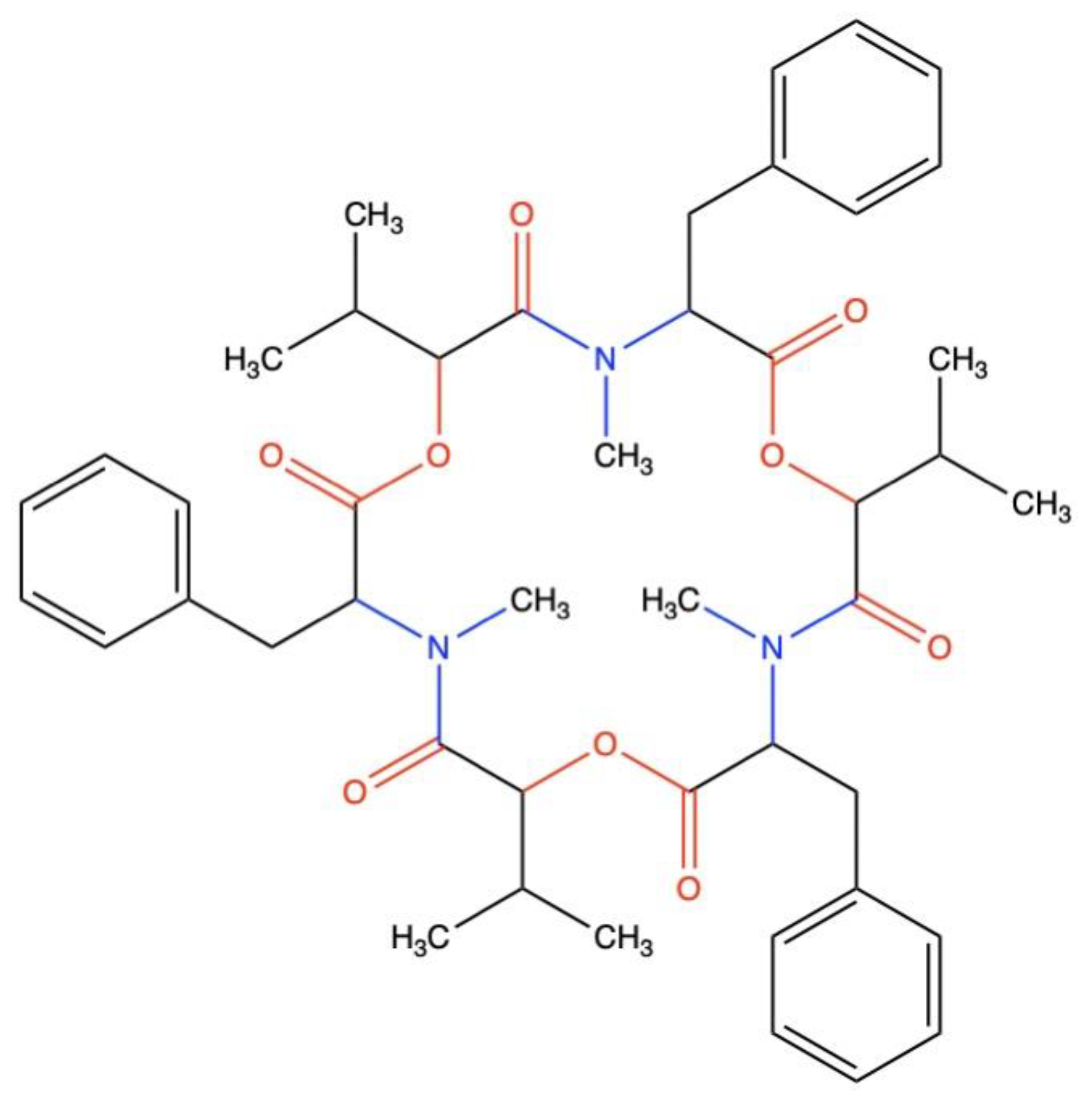
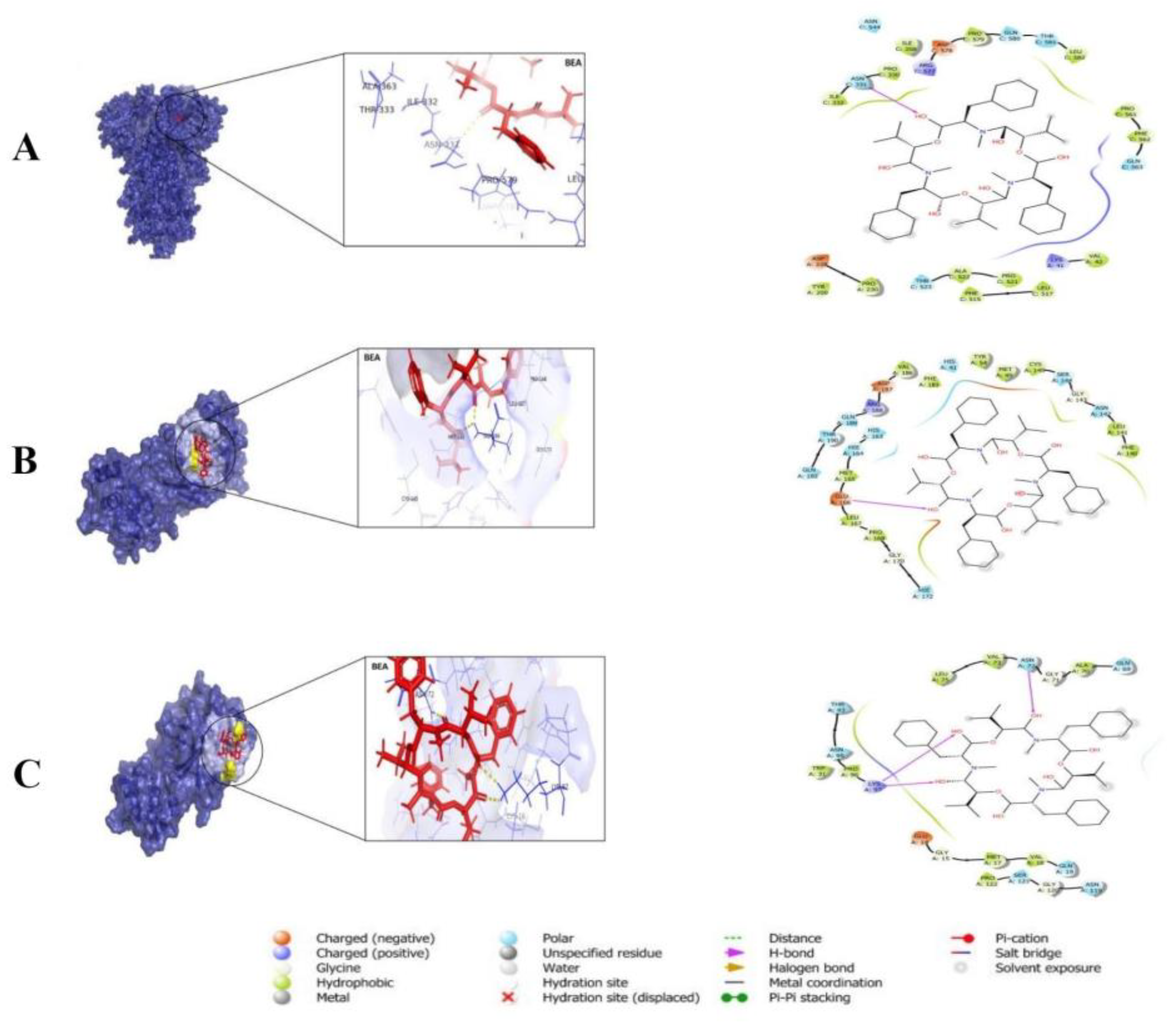
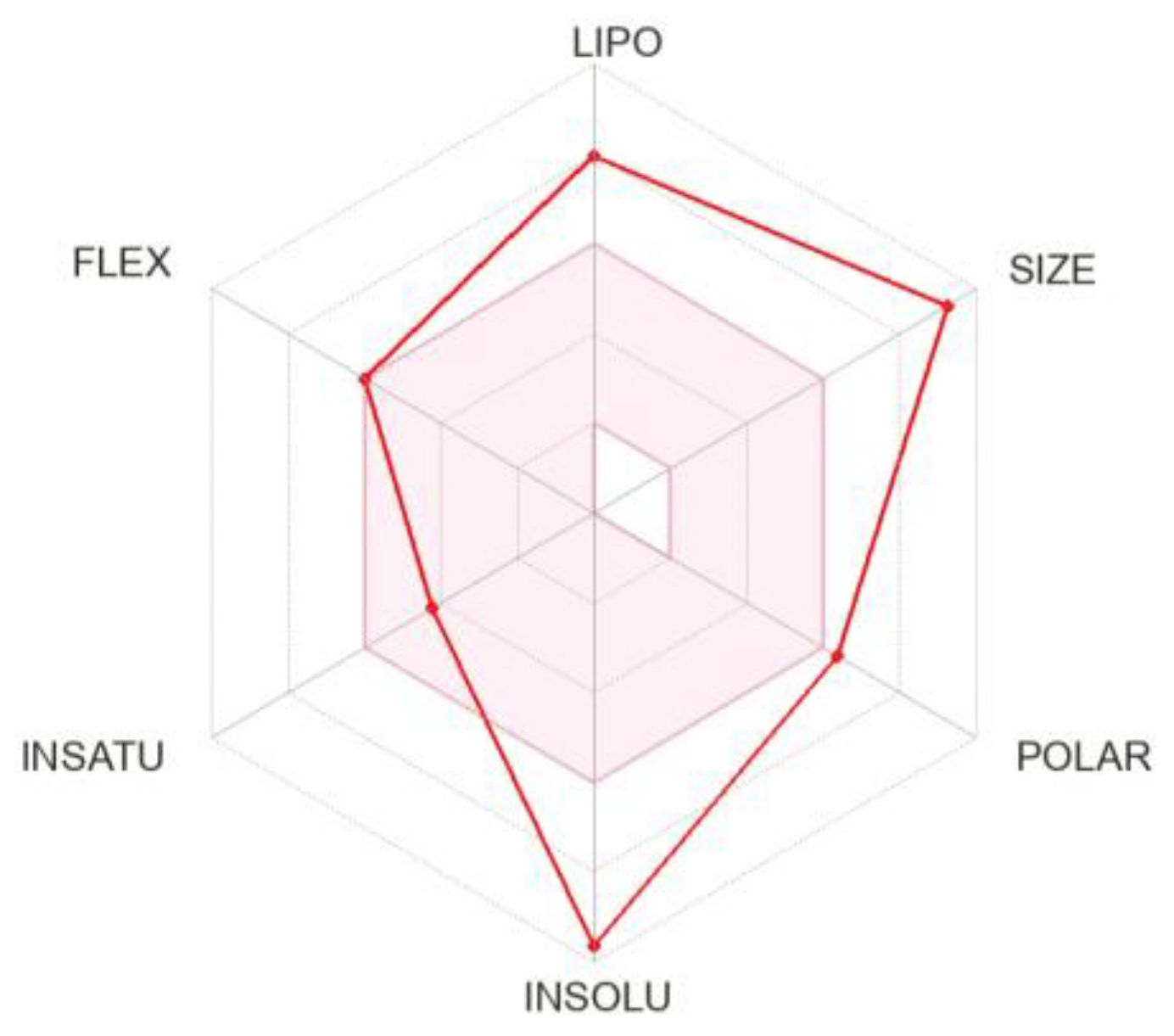
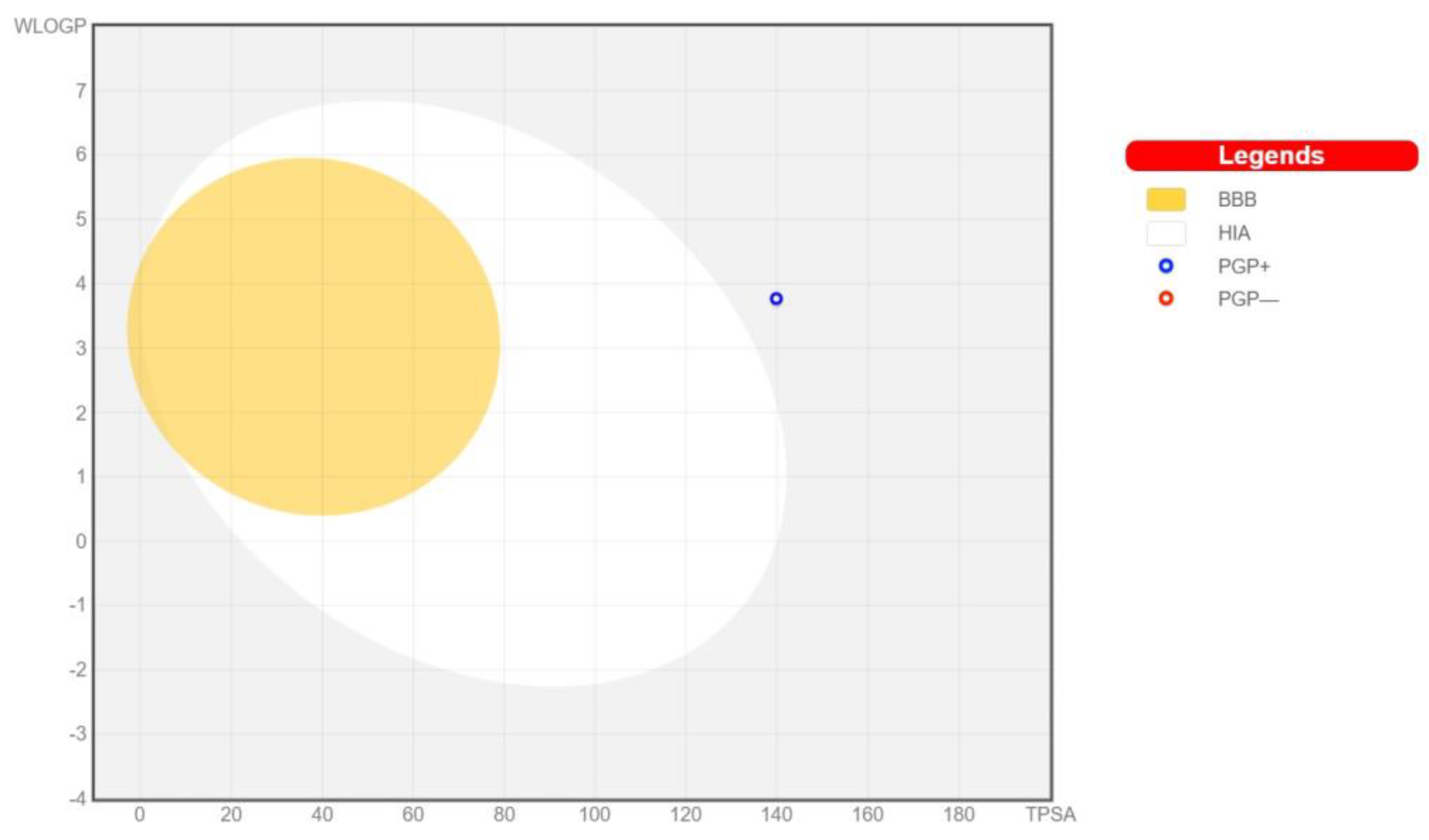
3.3. Pharmacokinetics and Drug-Likeness of BEA
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barrios-González, J. Secondary metabolites production: Physiological advantages in solid-state fermentation. Curr. Dev. Biotechnol. Bioeng. 2018, 1, 257–283. [Google Scholar] [CrossRef]
- Mosunova, O.; Navarro-Muñoz, J.C.; Collemare, J. The Biosynthesis of Fungal Secondary Metabolites: From Fundamentals to Biotechnological Applications. In Encyclopedia of Mycology; Zaragoza, Ó., Casadevall, A., Eds.; Elsevier: Oxford, UK, 2021; pp. 458–476. [Google Scholar]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar] [CrossRef]
- Rehner, S.A.; Buckley, E. A Beauveria phylogeny inferred from nuclear ITS and EF1-α sequences: Evidence for cryptic diversification and links to Cordyceps teleomorphs. Mycologia 2005, 97, 84–98. [Google Scholar] [CrossRef] [PubMed]
- De Faria, M.R.; Wraight, S.P. Mycoinsecticides and mycoacaricides: A comprehensive list with worldwide coverage and international classification of formulation types. Biol. Control 2007, 43, 237–256. [Google Scholar] [CrossRef]
- Sung, J.; Lee, J.; Humber, R.A.; Sung, G.; Shrestha, B. Cordyceps bassiana and production of stromata in vitro showing Beauveria anamorph in Korea. Mycobiology 2006, 34, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Rehner, S.A.; Minnis, A.M.; Sung, G.; Luangsa-ard, J.J.; Devotto, L.; Humber, R.A. Phylogeny and systematics of the anamorphic, entomopathogenic genus Beauveria. Mycologia 2011, 103, 1055–1073. [Google Scholar] [CrossRef] [PubMed]
- Chandler, D.; Davidson, G.; Jacobson, R.J. Laboratory and glasshouse evaluation of entomopathogenic fungi against the two-spotted spider mite, Tetranychus urticae (Acari: Tetranychidae), on tomato, Lycopersicon esculentum. Biocontrol Sci. Technol. 2005, 15, 37–54. [Google Scholar] [CrossRef]
- Immediato, D.; Camarda, A.; Iatta, R.; Puttilli, M.R.; Ramos, R.A.N.; Di Paola, G.; Giangaspero, A.; Otranto, D.; Cafarchia, C. Laboratory evaluation of a native strain of Beauveria bassiana for controlling Dermanyssus gallinae (De Geer, 1778) (Acari: Dermanyssidae). Vet. Parasitol. 2015, 212, 478–482. [Google Scholar] [CrossRef]
- Vestergaard, S.; Cherry, A.; Keller, S.; Goettel, M. Safety of hyphomycete fungi as microbial control agents. In Environmental Impacts of Microbial Insecticides: Need and Methods for Risk Assessment; Springer: Dordrecht, The Netherlands, 2003; pp. 35–62. [Google Scholar]
- Castellanos-Moguel, J.; Mier, T.; Reyes-Montes, M.d.R.; Navarro Barranco, H.; Zepeda Rodríguez, A.; Pérez-Torres, A.; Toriello, C. Fungal growth development index and ultrastructural study of whiteflies infected by three Isaria fumosorosea isolates of different pathogenicity. Rev. Mex. De Micol. 2013, 38, 23–33. [Google Scholar]
- Feng, M.G.; Poprawski, T.J.; Khachatourians, G.G. Production, formulation and application of the entomopathogenic fungus Beauveria bassiana for insect control: Current status. Biocontrol Sci. Technol. 1994, 4, 3–34. [Google Scholar] [CrossRef]
- Pendland, J.C.; Hung, S.Y.; Boucias, D.G. Evasion of host defense by in vivo-produced protoplast-like cells of the insect mycopathogen Beauveria bassiana. J. Bacteriol. 1993, 175, 5962–5969. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xu, L. Beauvericin, a bioactive compound produced by fungi: A short review. Molecules 2012, 17, 2367–2377. [Google Scholar] [CrossRef]
- Wu, Q.; Patocka, J.; Nepovimova, E.; Kuca, K. A review on the synthesis and bioactivity aspects of beauvericin, a Fusarium mycotoxin. Front. Pharmacol. 2018, 9, 1338. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Orozco, R.; Wijeratne, E.K.; Gunatilaka, A.L.; Stock, S.P.; Molnár, I. Biosynthesis of the cyclooligomer depsipeptide beauvericin, a virulence factor of the entomopathogenic fungus Beauveria bassiana. Chem. Biol. 2008, 15, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Mallebrera, B.; Prosperini, A.; Font, G.; Ruiz, M.J. In vitro mechanisms of Beauvericin toxicity: A review. Food Chem. Toxicol. 2018, 111, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.M.; Donzelli, B.G.; Krasnoff, S.B.; Keyhani, N.O. Discovering the secondary metabolite potential encoded within entomopathogenic fungi. Nat. Prod. Rep. 2014, 31, 1287–1305. [Google Scholar] [CrossRef]
- Ojcius, D.M.; Zychlinsky, A.; Zheng, L.M.; Young, J.D. Ionophore-induced apoptosis: Role of DNA fragmentation and calcium fluxes. Exp. Cell Res. 1991, 197, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Kouri, K.; Lemmens, M.; Lemmens-Gruber, R. Beauvericin-induced channels in ventricular myocytes and liposomes. Biochim. Biophys. Acta (BBA)-Biomembr. 2003, 1609, 203–210. [Google Scholar] [CrossRef]
- Jow, G.; Chou, C.; Chen, B.; Tsai, J. Beauvericin induces cytotoxic effects in human acute lymphoblastic leukemia cells through cytochrome c release, caspase 3 activation: The causative role of calcium. Cancer Lett. 2004, 216, 165–173. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Keyhani, N.O.; Xia, Y.; Cao, Y. The regulatory role of the transcription factor Crz1 in stress tolerance, pathogenicity, and its target gene expression in Metarhizium acridum. Appl. Microbiol. Biotechnol. 2017, 101, 5033–5043. [Google Scholar] [CrossRef] [PubMed]
- Pócsfalvi, G.; Di Landa, G.; Ferranti, P.; Ritieni, A.; Randazzo, G.; Malorni, A. Observation of non-covalent interactions between beauvericin and oligonucleotides using electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 1997, 11, 265–272. [Google Scholar] [CrossRef]
- Grove, J.F.; Pople, M. The insecticidal activity of beauvericin and the enniatin complex. Mycopathologia 1980, 70, 103–105. [Google Scholar] [CrossRef]
- Göldel, B.; Lemic, D.; Bažok, R. Alternatives to synthetic insecticides in the control of the colorado potato beetle (Leptinotarsa decemlineata Say) and their environmental benefits. Agriculture 2020, 10, 611. [Google Scholar] [CrossRef]
- Ganassi, S.; Moretti, A.; Pagliai, A.M.B.; Logrieco, A.; Sabatini, M.A. Effects of beauvericin on Schizaphis graminum (Aphididae). J. Invertebr. Pathol. 2002, 80, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Fornelli, F.; Minervini, F.; Logrieco, A. Cytotoxicity of fungal metabolites to lepidopteran (Spodoptera frugiperda) cell line (SF-9). J. Invertebr. Pathol. 2004, 85, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Leland, J.E.; McGuire, M.R.; Grace, J.A.; Jaronski, S.T.; Ulloa, M.; Park, Y.; Plattner, R.D. Strain selection of a fungal entomopathogen, Beauveria bassiana, for control of plant bugs (Lygus spp.) (Heteroptera: Miridae). Biol. Control 2005, 35, 104–114. [Google Scholar] [CrossRef]
- Al Khoury, C.; Guillot, J.; Nemer, N. Lethal activity of beauvericin, a Beauveria bassiana mycotoxin, against the two-spotted spider mites, Tetranychus urticae Koch. J. Appl. Entomol. 2019, 143, 974–983. [Google Scholar] [CrossRef]
- Zhang, L.; Yan, K.; Zhang, Y.; Huang, R.; Bian, J.; Zheng, C.; Sun, H.; Chen, Z.; Sun, N.; An, R. High-throughput synergy screening identifies microbial metabolites as combination agents for the treatment of fungal infections. Proc. Natl. Acad. Sci. USA 2007, 104, 4606–4611. [Google Scholar] [CrossRef]
- Shekhar-Guturja, T.; Gunaherath, G.K.B.; Wijeratne, E.K.; Lambert, J.; Averette, A.F.; Lee, S.C.; Kim, T.; Bahn, Y.; Tripodi, F.; Ammar, R. Dual action antifungal small molecule modulates multidrug efflux and TOR signaling. Nat. Chem. Biol. 2016, 12, 867–875. [Google Scholar] [CrossRef]
- Al Khoury, C.; Nemer, N.; Nemer, G. Beauvericin potentiates the activity of pesticides by neutralizing the ATP-binding cassette transporters in arthropods. Sci. Rep. 2021, 11, 10865. [Google Scholar] [CrossRef] [PubMed]
- Osagie, A.E.; Olalekan, S.H. Multiple Drug resistance: A fast-growing threat. Bio. Med. 2019, 21, 15715–15726. [Google Scholar]
- Llor, C.; Bjerrum, L. Antimicrobial resistance: Risk associated with antibiotic overuse and initiatives to reduce the problem. Ther. Adv. Drug Saf. 2014, 5, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Cho, I.H.; Jeong, B.C.; Lee, S.H. Strategies to minimize antibiotic resistance. Int. J. Environ. Res. Public Health 2013, 10, 4274–4305. [Google Scholar] [CrossRef] [PubMed]
- Chitsaz, M.; Brown, M.H. The role played by drug efflux pumps in bacterial multidrug resistance. Essays Biochem. 2017, 61, 127–139. [Google Scholar] [PubMed]
- Merzendorfer, H. ABC transporters and their role in protecting insects from pesticides and their metabolites. In Advances in Insect Physiology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 46, pp. 1–72. [Google Scholar]
- Lage, H. ABC-transporters: Implications on drug resistance from microorganisms to human cancers. Int. J. Antimicrob. Agents 2003, 22, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Liu, M.; Zhang, Y.; Liu, X.; Huang, R.; Song, F.; Dai, H.; Ren, B.; Sun, N.; Pei, G. Beauvericin counteracted multi-drug resistant Candida albicans by blocking ABC transporters. Synth. Syst. Biotechnol. 2016, 1, 158–168. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- E Lohning, A.; M Levonis, S.; Williams-Noonan, B.; S Schweiker, S. A practical guide to molecular docking and homology modelling for medicinal chemists. Curr. Top. Med. Chem. 2017, 17, 2023–2040. [Google Scholar] [CrossRef]
- Saikia, S.; Bordoloi, M. Molecular docking: Challenges, advances and its use in drug discovery perspective. Curr. Drug Targets 2019, 20, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Al Khoury, C.; Thoumi, S.; Tokajian, S.; Sinno, A.; Nemer, G.; El Beyrouthy, M.; Rahy, K. ABC transporter inhibition by beauvericin partially overcomes drug resistance in Leishmania tropica. Antimicrob. Agents Chemother. 2024, e01368-23. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.; An, D.; Song, H.; Lee, C. Beauvericin and enniatins H, I and MK1688 are new potent inhibitors of human immunodeficiency virus type-1 integrase. J. Antibiot. 2009, 62, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Fesen, M.R.; Pommier, Y.; Leteurtre, F.; Hiroguchi, S.; Yung, J.; Kohn, K.W. Inhibition of HIV-1 integrase by flavones, caffeic acid phenethyl ester (CAPE) and related compounds. Biochem. Pharmacol. 1994, 48, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.; Park, J.Y.; Lee, J.; Yoo, H.H.; Hahm, D.; Lee, S.C.; Lee, S.; Hwang, G.S.; Jung, K.; Kang, K.S. Anti-inflammatory effects and corresponding mechanisms of cirsimaritin extracted from Cirsium japonicum var. maackii Maxim. Bioorg. Med. Chem. Lett. 2017, 27, 3076–3080. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C.; Goldstein, S.A.; Rasmussen, A.L.; Robertson, D.L.; Crits-Christoph, A.; Wertheim, J.O.; Anthony, S.J.; Barclay, W.S.; Boni, M.F.; Doherty, P.C. The origins of SARS-CoV-2: A critical review. Cell 2021, 184, 4848–4856. [Google Scholar] [CrossRef] [PubMed]
- Adil, M.T.; Rahman, R.; Whitelaw, D.; Jain, V.; Al-Taan, O.; Rashid, F.; Munasinghe, A.; Jambulingam, P. SARS-CoV-2 and the pandemic of COVID-19. Postgrad. Med. J. 2021, 97, 110–116. [Google Scholar] [CrossRef]
- Wu, D.; Wu, T.; Liu, Q.; Yang, Z. The SARS-CoV-2 outbreak: What we know. Int. J. Infect. Dis. 2020, 94, 44–48. [Google Scholar] [CrossRef]
- Alsulami, A.F.; Thomas, S.E.; Jamasb, A.R.; Beaudoin, C.A.; Moghul, I.; Bannerman, B.; Copoiu, L.; Vedithi, S.C.; Torres, P.; Blundell, T.L. SARS-CoV-2 3D database: Understanding the coronavirus proteome and evaluating possible drug targets. Brief. Bioinform. 2021, 22, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Vicenti, I.; Zazzi, M.; Saladini, F. SARS-CoV-2 RNA-dependent RNA polymerase as a therapeutic target for COVID-19. Expert Opin. Ther. Pat. 2021, 31, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, K.S.; Ansari, M.; Moghaddam, M.S.H.; Ebrahimi, Z.; Shahlaei, M.; Moradi, S. In silico investigation on the inhibitory effect of fungal secondary metabolites on RNA dependent RNA polymerase of SARS-CoV-II: A docking and molecular dynamic simulation study. Comput. Biol. Med. 2021, 135, 104613. [Google Scholar] [CrossRef] [PubMed]
- Pathania, S.; Rawal, R.K.; Singh, P.K. RdRp (RNA-dependent RNA polymerase): A key target providing anti-virals for the management of various viral diseases. J. Mol. Struct. 2022, 1250, 131756. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar] [PubMed]
- Lindahl, E.; Hess, B.; Van Der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. Mol. Model. Annu. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Al Khoury, C.; Bashir, Z.; Tokajian, S.; Nemer, N.; Merhi, G.; Nemer, G. In silico evidence of beauvericin antiviral activity against SARS-CoV-2. Comput. Biol. Med. 2021, 141, 105171. [Google Scholar] [CrossRef]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef]
- Xie, L.; Xie, L.; Kinnings, S.L.; Bourne, P.E. Novel computational approaches to polypharmacology as a means to define responses to individual drugs. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Amporndanai, K.; Meng, X.; Shang, W.; Jin, Z.; Rogers, M.; Zhao, Y.; Rao, Z.; Liu, Z.; Yang, H.; Zhang, L. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 2021, 12, 3061. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Reichel, A.; Lienau, P. Pharmacokinetics in drug discovery: An exposure-centred approach to optimising and predicting drug efficacy and safety. In New Approaches to Drug Discovery; Springer: Cham, Switzerland, 2015; pp. 235–260. [Google Scholar]
- Ruiz-Garcia, A.; Bermejo, M.; Moss, A.; Casabo, V.G. Pharmacokinetics in drug discovery. J. Pharm. Sci. 2008, 97, 654–690. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of octanol− water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple method of calculating octanol/water partition coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef]
- Moriguchi, I.; Hirono, S.; Nakagome, I.; Hirano, H. Comparison of reliability of log P values for drugs calculated by several methods. Chem. Pharm. Bull. 1994, 42, 976–978. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef] [PubMed]
- Jestoi, M. Emerging Fusarium-mycotoxins fusaproliferin, beauvericin, enniatins, and moniliformin—A review. Crit. Rev. Food Sci. Nutr. 2008, 48, 21–49. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.A.; Smith, J.S. Liquid chromatography/thermospray/mass spectrometry analysis of beauvericin. J. Agric. Food Chem. 1997, 45, 1234–1239. [Google Scholar] [CrossRef]
- Rodríguez-Carrasco, Y.; Heilos, D.; Richter, L.; Süssmuth, R.D.; Heffeter, P.; Sulyok, M.; Kenner, L.; Berger, W.; Dornetshuber-Fleiss, R. Mouse tissue distribution and persistence of the food-born fusariotoxins Enniatin B and Beauvericin. Toxicol. Lett. 2016, 247, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Braden, B.; Hamilton, J.A.; Sabesan, M.N.; Steinrauf, L.K. Crystal structure of a beauvericin-barium picrate complex. J. Am. Chem. Soc. 1980, 102, 2704–2709. [Google Scholar] [CrossRef]
- Khaled, M.A.; Davies, D.B. Solution and ion-complexed conformations of beauvericin determined by proton magnetic resonance spectroscopy. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 1982, 704, 186–196. [Google Scholar] [CrossRef]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the general solubility equation: In silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [CrossRef]
- Yalkowsky, S.H.; Valvani, S.C. Solubility and partitioning I: Solubility of nonelectrolytes in water. J. Pharm. Sci. 1980, 69, 912–922. [Google Scholar] [CrossRef]
- Hamill, R.L.; Higgens, C.E.; Boaz, H.E.; Gorman, M. The structure op beauvericin, a new depsipeptide antibiotic toxic to Artemia salina. Tetrahedron Lett. 1969, 10, 4255–4258. [Google Scholar] [CrossRef]
- Logrieco, A.; Moretti, A.; Ritieni, A.; Caiaffa, M.F.; Macchia, L. Beauvericin: Chemistry, biology and significance. In Advances in Microbial Toxin Research and Its Biotechnological Exploitation; Springer: Boston, MA, USA, 2002; pp. 23–30. [Google Scholar]
- Lombardo, F.; Desai, P.V.; Arimoto, R.; Desino, K.E.; Fischer, H.; Keefer, C.E.; Petersson, C.; Winiwarter, S.; Broccatelli, F. In Silico absorption, distribution, metabolism, excretion, and pharmacokinetics (ADME-PK): Utility and best practices. an industry perspective from the international consortium for innovation through quality in pharmaceutical development: Miniperspective. J. Med. Chem. 2017, 60, 9097–9113. [Google Scholar] [CrossRef] [PubMed]
- Devreese, M.; De Baere, S.; De Backer, P.; Croubels, S. Quantitative determination of the Fusarium mycotoxins beauvericin, enniatin A, A1, B and B1 in pig plasma using high performance liquid chromatography–tandem mass spectrometry. Talanta 2013, 106, 212–219. [Google Scholar] [CrossRef]
- Prosperini, A.; Meca, G.; Font, G.; Ruiz, M.J. Study of the cytotoxic activity of beauvericin and fusaproliferin and bioavailability in vitro on Caco-2 cells. Food Chem. Toxicol. 2012, 50, 2356–2361. [Google Scholar] [CrossRef]
- Meca, G.; Mañes, J.; Font, G.; Ruiz, M.J. Study of the potential toxicity of enniatins A, A1, B, B1 by evaluation of duodenal and colonic bioavailability applying an in vitro method by Caco-2 cells. Toxicon 2012, 59, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Taevernier, L.; Bracke, N.; Veryser, L.; Wynendaele, E.; Gevaert, B.; Peremans, K.; De Spiegeleer, B. Blood-brain barrier transport kinetics of the cyclic depsipeptide mycotoxins beauvericin and enniatins. Toxicol. Lett. 2016, 258, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.; Neff, A.P. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar]
- Huang, S.; Strong, J.M.; Zhang, L.; Reynolds, K.S.; Nallani, S.; Temple, R.; Abraham, S.; Habet, S.A.; Baweja, R.K.; Burckart, G.J. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J. Clin. Pharmacol. 2008, 48, 662–670. [Google Scholar] [CrossRef]
- Mei, L.; Zhang, L.; Dai, R. An inhibition study of beauvericin on human and rat cytochrome P450 enzymes and its pharmacokinetics in rats. J. Enzym. Inhib. Med. Chem. 2009, 24, 753–762. [Google Scholar] [CrossRef]
- Potts, R.O.; Guy, R.H. Predicting skin permeability. Pharm. Res. 1992, 9, 663–669. [Google Scholar] [CrossRef]
- Taevernier, L.; Veryser, L.; Roche, N.; Peremans, K.; Burvenich, C.; Delesalle, C.; De Spiegeleer, B. Human skin permeation of emerging mycotoxins (beauvericin and enniatins). J. Expo. Sci. Environ. Epidemiol. 2016, 26, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Demain, A.L. Pharmaceutically active secondary metabolites of microorganisms. Appl. Microbiol. Biotechnol. 1999, 52, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wilkinson, B. Drug discovery beyond the ‘rule-of-five’. Curr. Opin. Biotechnol. 2007, 18, 478–488. [Google Scholar] [CrossRef]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein–protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physicochemical Properties | |
|---|---|
| Formula | C45H57N3O9 |
| Molecular weight | 783.95 g/mol |
| Num. heavy atoms | 57 |
| Num. atom. heavy atoms | 18 |
| Fraction Csp3 | 0.47 |
| Num. rotatable bonds | 9 |
| Num. H-bond acceptors | 9 |
| Num. H-bond donors | 0 |
| Molar Refractivity | 228.14 |
| TPSA | 139.83 Å2 |
| Lipophilicity | |
|---|---|
| Log Po/w (iLOGP) | 5.29 |
| Log Po/w (XLOGP3) | 8.42 |
| Log Po/w (WLOGP) | 3.77 |
| Log Po/w (MLOGP) | 3.14 |
| Log Po/w (SILICOS-IT) | 5.43 |
| Consensus Log Po/w | 5.21 |
| Water Solubility | |
|---|---|
| Log S (ESOL) | −9.64 |
| Solubility | 1.78 × 10−07 mg/mL; 2.27 × 10−10 mol/L |
| Class | Poorly soluble |
| Log S (Ali) | −11.23 |
| Solubility | 4.67 × 10−09 mg/mL; 5.96 × 10−12 mol/L |
| Class | Insoluble |
| Log S (SILICOS-IT) | −10.22 |
| Solubility | 4.68 × 10−08 mg/mL; 5.97 × 10−11 mol/L |
| Class | Insoluble |
| Pharmacokinetics | |
|---|---|
| GI absorption | Low |
| BBB-permeant | No |
| P-gp substrate | Yes |
| CYP1A2 inhibitor | No |
| CYP2C19 inhibitor | No |
| CYP2C9 inhibitor | No |
| CYP2D6 inhibitor | No |
| CYP3A4 inhibitor | No |
| Log Kp (skin permeation) | −5.10 cm/s |
| Druglikeness | |
|---|---|
| Lipinski | No; 2 violations: MW > 500, NorO > 10 |
| Ghose | No; 3 violations: MW > 480, MR > 130, #atoms > 70 |
| Veber | Yes |
| Egan | No; 1 violation: TPSA > 131.6 |
| Muegge | No; 2 violations: MW > 600, XLOGP3 > 5 |
| Bioavailability Score | 0.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Khoury, C.; Tokajian, S.; Nemer, N.; Nemer, G.; Rahy, K.; Thoumi, S.; Al Samra, L.; Sinno, A. Computational Applications: Beauvericin from a Mycotoxin into a Humanized Drug. Metabolites 2024, 14, 232. https://doi.org/10.3390/metabo14040232
Al Khoury C, Tokajian S, Nemer N, Nemer G, Rahy K, Thoumi S, Al Samra L, Sinno A. Computational Applications: Beauvericin from a Mycotoxin into a Humanized Drug. Metabolites. 2024; 14(4):232. https://doi.org/10.3390/metabo14040232
Chicago/Turabian StyleAl Khoury, Charbel, Sima Tokajian, Nabil Nemer, Georges Nemer, Kelven Rahy, Sergio Thoumi, Lynn Al Samra, and Aia Sinno. 2024. "Computational Applications: Beauvericin from a Mycotoxin into a Humanized Drug" Metabolites 14, no. 4: 232. https://doi.org/10.3390/metabo14040232
APA StyleAl Khoury, C., Tokajian, S., Nemer, N., Nemer, G., Rahy, K., Thoumi, S., Al Samra, L., & Sinno, A. (2024). Computational Applications: Beauvericin from a Mycotoxin into a Humanized Drug. Metabolites, 14(4), 232. https://doi.org/10.3390/metabo14040232