

MIGRENE: The Toolbox for Microbial and Individualized GEMs, Reactobiome and Community Network Modelling

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Method
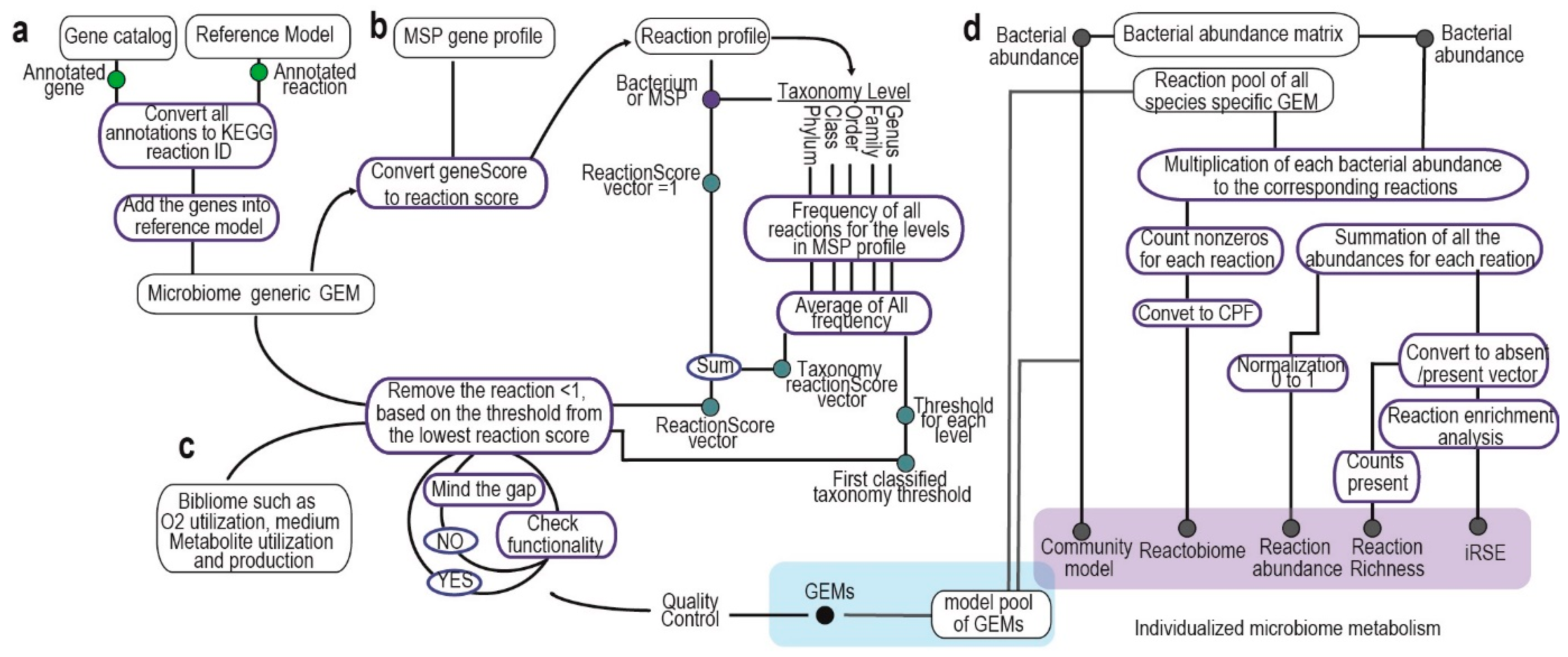
2.1. Integration of Bacterial Gene Catalog into Metabolic Model and Microbiome Reference GEM Generation
2.2. Calculation of Reaction Score and Taxonomy Threshold
2.3. Species-Specific Genome-Scale Metabolic Model Generation
2.4. Generation of Individualized Metabolic Microbiome: Reaction Richness, Reaction Abundance and Reactobiome, iRSE and Community Models
2.5. Processing and Downstream Analysis of Liver Cirrhosis Metagenomics
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Brien, E.J.; Monk, J.M.; Palsson, B.O. Using Genome-scale Models to Predict Biological Capabilities. Cell 2015, 161, 971–987. [Google Scholar] [CrossRef] [PubMed]
- Magnusdottir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jager, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef]
- Tramontano, M.; Andrejev, S.; Pruteanu, M.; Klunemann, M.; Kuhn, M.; Galardini, M.; Jouhten, P.; Zelezniak, A.; Zeller, G.; Bork, P.; et al. Nutritional preferences of human gut bacteria reveal their metabolic idiosyncrasies. Nat. Microbiol. 2018, 3, 514–522. [Google Scholar] [CrossRef]
- Heinken, A.; Hertel, J.; Acharya, G.; Ravcheev, D.A.; Nyga, M.; Okpala, O.E.; Hogan, M.; Magnusdottir, S.; Martinelli, F.; Nap, B.; et al. Genome-scale metabolic reconstruction of 7302 human microorganisms for personalized medicine. Nat. Biotechnol. 2023, 41, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 2021, 49, D575–D588. [Google Scholar] [CrossRef]
- Gallemann, D.; Eyer, P. Formation of hydrogen peroxide during precipitation of red cells with perchloric acid. A cautionary note for precise determination of pyruvate, GSH, and NAD(P)H. Anal. Biochem. 1990, 191, 347–353. [Google Scholar] [CrossRef]
- Chan, S.H.J.; Simons, M.N.; Maranas, C.D. SteadyCom: Predicting microbial abundances while ensuring community stability. PLoS Comput. Biol. 2017, 13, e1005539. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, R.A.; Olivier, B.G.; Roling, W.F.; Teusink, B.; Bruggeman, F.J. Community flux balance analysis for microbial consortia at balanced growth. PLoS ONE 2013, 8, e64567. [Google Scholar] [CrossRef]
- Zhuang, K.; Izallalen, M.; Mouser, P.; Richter, H.; Risso, C.; Mahadevan, R.; Lovley, D.R. Genome-scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. ISME J. 2011, 5, 305–316. [Google Scholar] [CrossRef]
- Succurro, A.; Segre, D.; Ebenhoh, O. Emergent Subpopulation Behavior Uncovered with a Community Dynamic Metabolic Model of Escherichia coli Diauxic Growth. mSystems 2019, 4, 1110–1128. [Google Scholar] [CrossRef]
- Brunner, J.D.; Chia, N. Minimizing the number of optimizations for efficient community dynamic flux balance analysis. PLoS Comput. Biol. 2020, 16, e1007786. [Google Scholar] [CrossRef]
- Dukovski, I.; Bajic, D.; Chacon, J.M.; Quintin, M.; Vila, J.C.C.; Sulheim, S.; Pacheco, A.R.; Bernstein, D.B.; Riehl, W.J.; Korolev, K.S.; et al. A metabolic modeling platform for the computation of microbial ecosystems in time and space (COMETS). Nat. Protoc. 2021, 16, 5030–5082. [Google Scholar] [CrossRef] [PubMed]
- Bauer, E.; Zimmermann, J.; Baldini, F.; Thiele, I.; Kaleta, C. BacArena: Individual-based metabolic modeling of heterogeneous microbes in complex communities. PLoS Comput. Biol. 2017, 13, e1005544. [Google Scholar] [CrossRef] [PubMed]
- Angeles-Martinez, L.; Hatzimanikatis, V. Spatio-temporal modeling of the crowding conditions and metabolic variability in microbial communities. PLoS Comput. Biol. 2021, 17, e1009140. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.T., Jr.; Benito-Vaquerizo, S.; Zimmermann, J.; Bajic, D.; Heinken, A.; Suarez-Diez, M.; Schaap, P.J. A structured evaluation of genome-scale constraint-based modeling tools for microbial consortia. PLoS Comput. Biol. 2023, 19, e1011363. [Google Scholar] [CrossRef] [PubMed]
- Zorrilla, F.; Buric, F.; Patil, K.R.; Zelezniak, A. metaGEM: Reconstruction of genome scale metabolic models directly from metagenomes. Nucleic Acids Res. 2021, 49, e126. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Mitchell, A.L.; Boland, M.; Forster, S.C.; Gloor, G.B.; Tarkowska, A.; Lawley, T.D.; Finn, R.D. A new genomic blueprint of the human gut microbiota. Nature 2019, 568, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 2019, 176, 649–662.e20. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Zheng, Z.; Shao, T.; Liu, L.; Xie, Z.; Le Chatelier, E.; He, Z.; Zhong, W.; Fan, Y.; Zhang, L.; et al. Correction to: Quantitative metagenomics reveals unique gut microbiome biomarkers in ankylosing spondylitis. Genome Biol. 2017, 18, 214. [Google Scholar] [CrossRef]
- Tierney, B.T.; Yang, Z.; Luber, J.M.; Beaudin, M.; Wibowo, M.C.; Baek, C.; Mehlenbacher, E.; Patel, C.J.; Kostic, A.D. The Landscape of Genetic Content in the Gut and Oral Human Microbiome. Cell Host. Microbe 2019, 26, 283–295.e8. [Google Scholar] [CrossRef]
- Ma, B.; France, M.T.; Crabtree, J.; Holm, J.B.; Humphrys, M.S.; Brotman, R.M.; Ravel, J. A comprehensive non-redundant gene catalog reveals extensive within-community intraspecies diversity in the human vagina. Nat. Commun. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Pons, N.; Batto, J.-M.; Kennedy, S.; Almeida, M.; Boumezbeur, F.; Moumen, B. METEOR, a platform for quantitative metagenomic profiling of complex ecosystems. In Proceedings of the JOBIM, Montpelier, France, 7–9 September 2010; pp. 7–9. [Google Scholar]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.Y.; Marshall, A.W.; Milsom, J.P.; Sherlock, S. Plasma amino-acid patterns in liver disease. Gut 1982, 23, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Amano, N.; Sato, S.; Kita, Y.; Ikeda, Y.; Kabemura, D.; Murata, A.; Yatagai, N.; Tsuzura, H.; Shimada, Y.; et al. Elevated serum tyrosine concentration is associated with a poor prognosis among patients with liver cirrhosis. Hepatol. Res. 2021, 51, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Proffitt, C.; Bidkhori, G.; Lee, S.; Tebani, A.; Mardinoglu, A.; Uhlen, M.; Moyes, D.L.; Shoaie, S. Genome-scale metabolic modelling of the human gut microbiome reveals changes in the glyoxylate and dicarboxylate metabolism in metabolic disorders. iScience 2022, 25, 104513. [Google Scholar] [CrossRef] [PubMed]
- Ezzamouri, B.; Rosario, D.; Bidkhori, G.; Lee, S.; Uhlen, M.; Shoaie, S. Metabolic modelling of the human gut microbiome in type 2 diabetes patients in response to metformin treatment. NPJ Syst. Biol. Appl. 2023, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Rosario, D.; Bidkhori, G.; Lee, S.; Bedarf, J.; Hildebrand, F.; Le Chatelier, E.; Uhlen, M.; Ehrlich, S.D.; Proctor, G.; Wullner, U.; et al. Systematic analysis of gut microbiome reveals the role of bacterial folate and homocysteine metabolism in Parkinson’s disease. Cell Rep. 2021, 34, 108807. [Google Scholar] [CrossRef] [PubMed]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef]
- Albertsen, M.; Hugenholtz, P.; Skarshewski, A.; Nielsen, K.L.; Tyson, G.W.; Nielsen, P.H. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 2013, 31, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Mannisto, V.; Farkkila, M.; Pussinen, P.; Jula, A.; Mannisto, S.; Lundqvist, A.; Valsta, L.; Salomaa, V.; Perola, M.; Aberg, F. Serum lipopolysaccharides predict advanced liver disease in the general population. JHEP Rep. 2019, 1, 345–352. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bidkhori, G.; Shoaie, S. MIGRENE: The Toolbox for Microbial and Individualized GEMs, Reactobiome and Community Network Modelling. Metabolites 2024, 14, 132. https://doi.org/10.3390/metabo14030132
Bidkhori G, Shoaie S. MIGRENE: The Toolbox for Microbial and Individualized GEMs, Reactobiome and Community Network Modelling. Metabolites. 2024; 14(3):132. https://doi.org/10.3390/metabo14030132
Chicago/Turabian StyleBidkhori, Gholamreza, and Saeed Shoaie. 2024. "MIGRENE: The Toolbox for Microbial and Individualized GEMs, Reactobiome and Community Network Modelling" Metabolites 14, no. 3: 132. https://doi.org/10.3390/metabo14030132
APA StyleBidkhori, G., & Shoaie, S. (2024). MIGRENE: The Toolbox for Microbial and Individualized GEMs, Reactobiome and Community Network Modelling. Metabolites, 14(3), 132. https://doi.org/10.3390/metabo14030132