
Metabolic Connectome and Its Role in the Prediction, Diagnosis, and Treatment of Complex Diseases



Abstract
1. Introduction
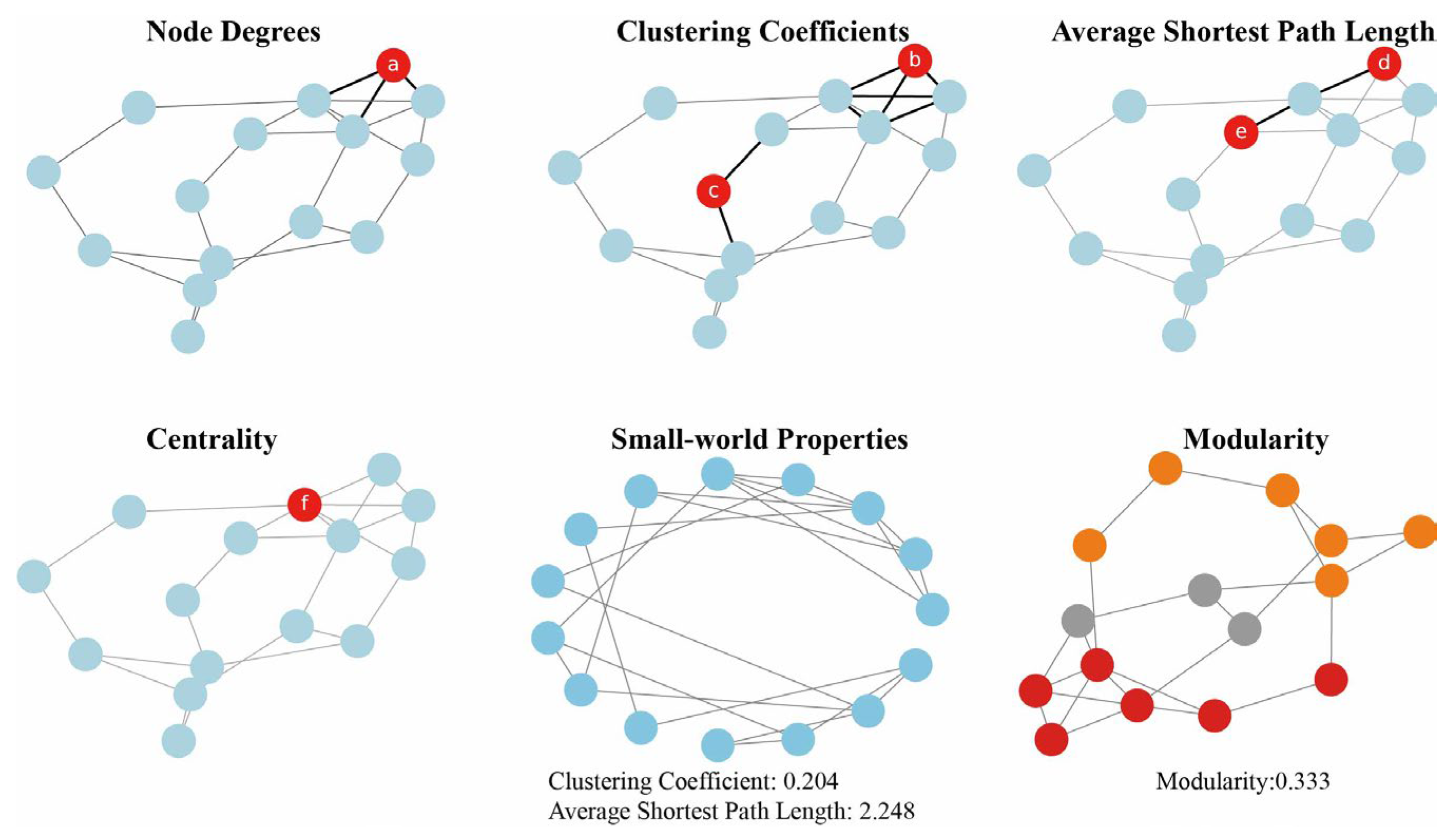
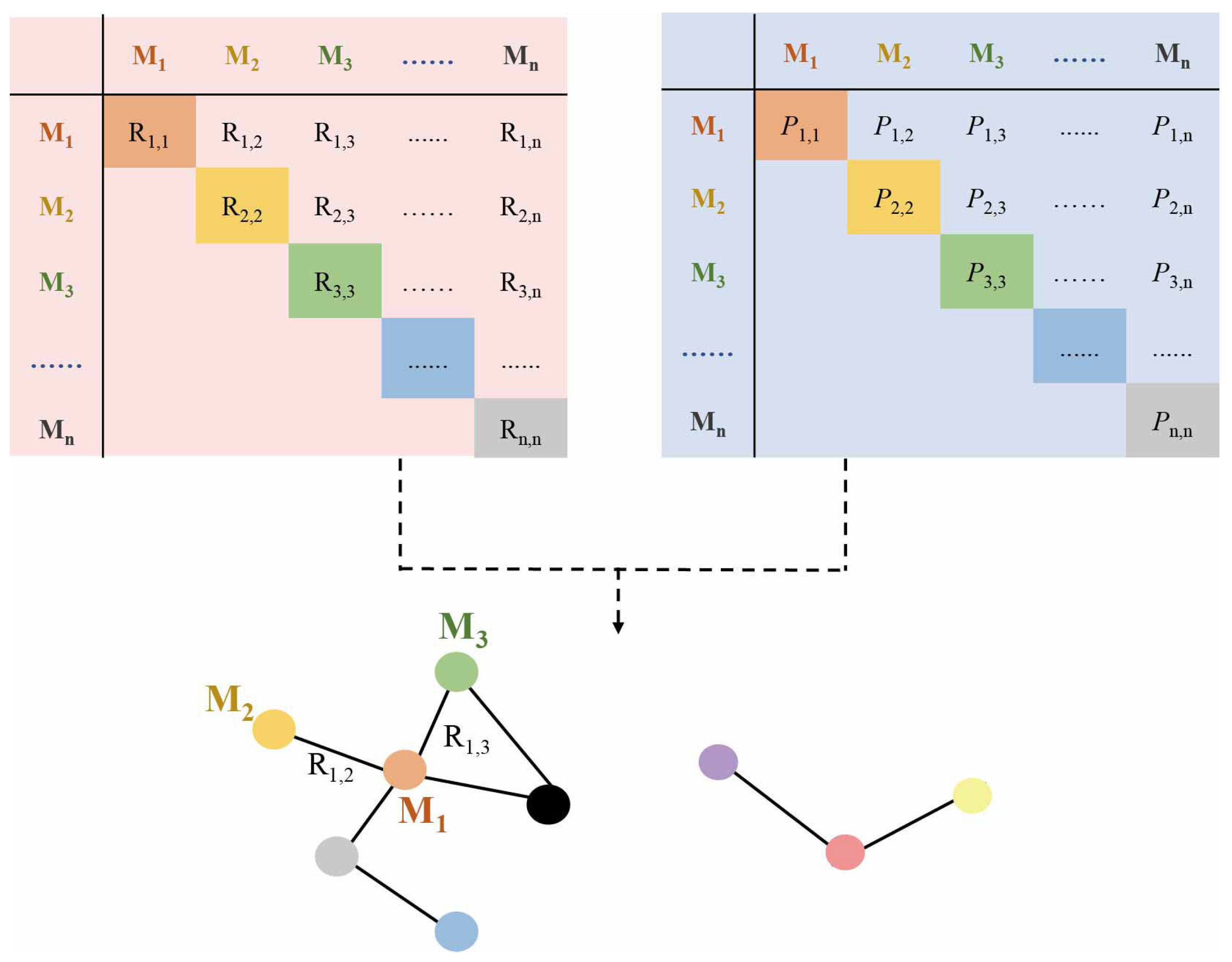
2. Construction Methods of Metabolic Networks
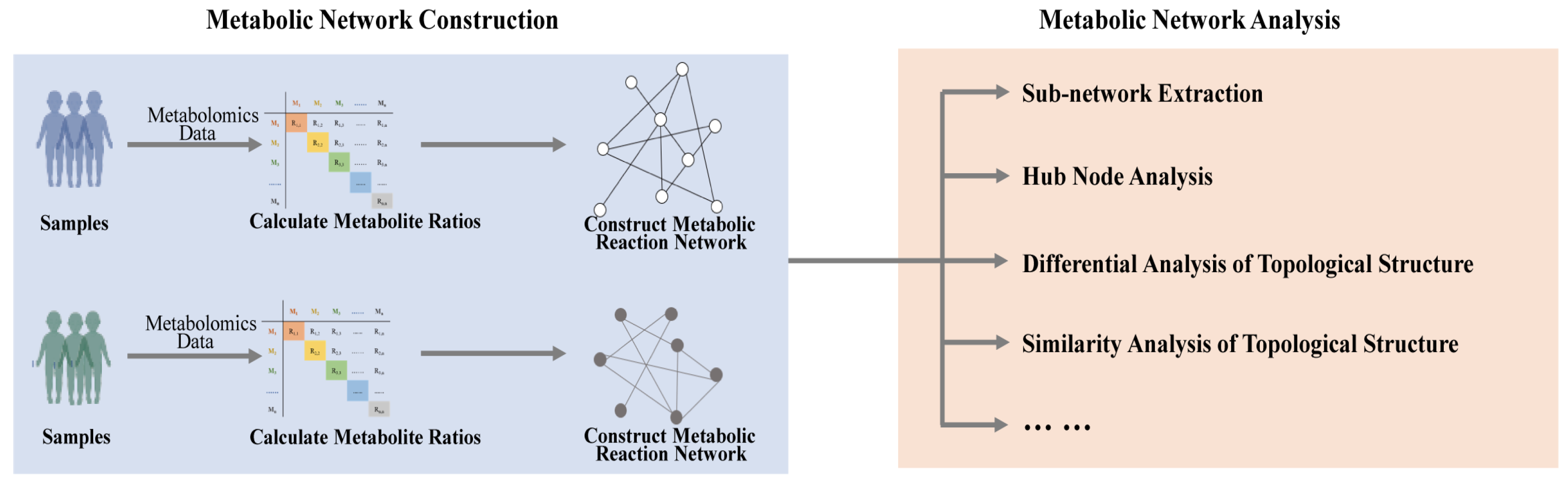
2.1. Correlation-Based Metabolic Network
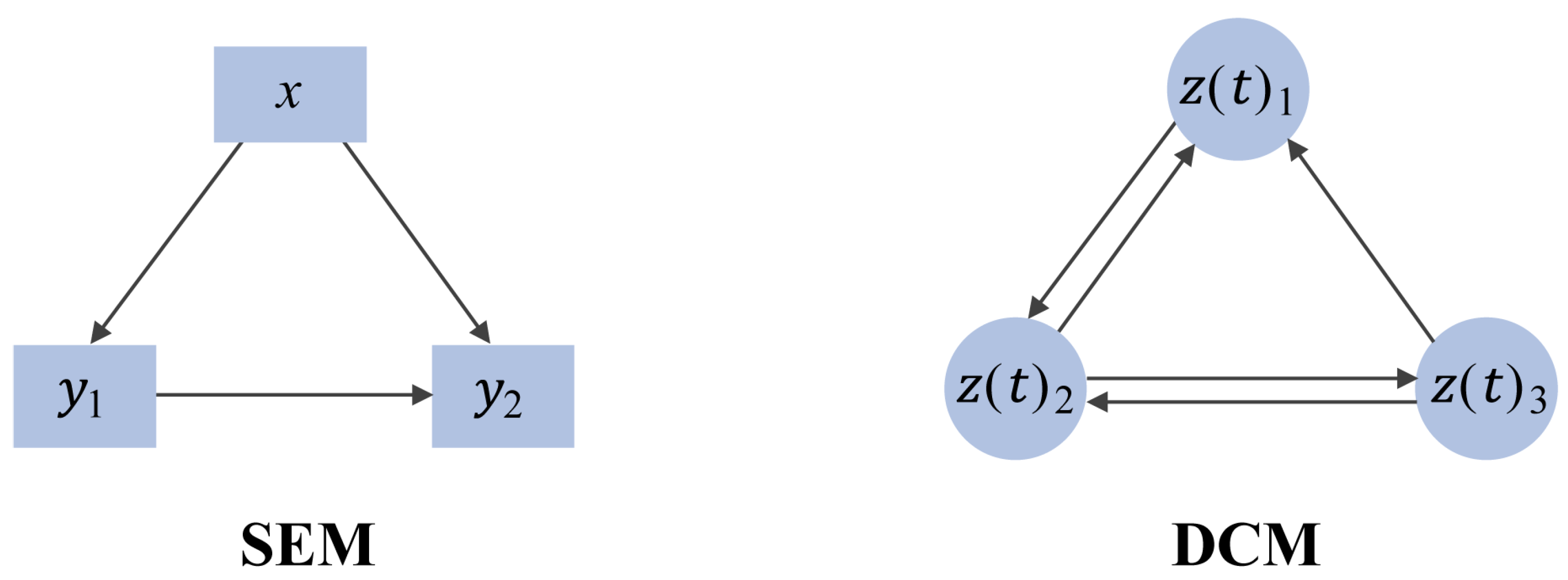
2.2. Causal-Based Metabolic Network
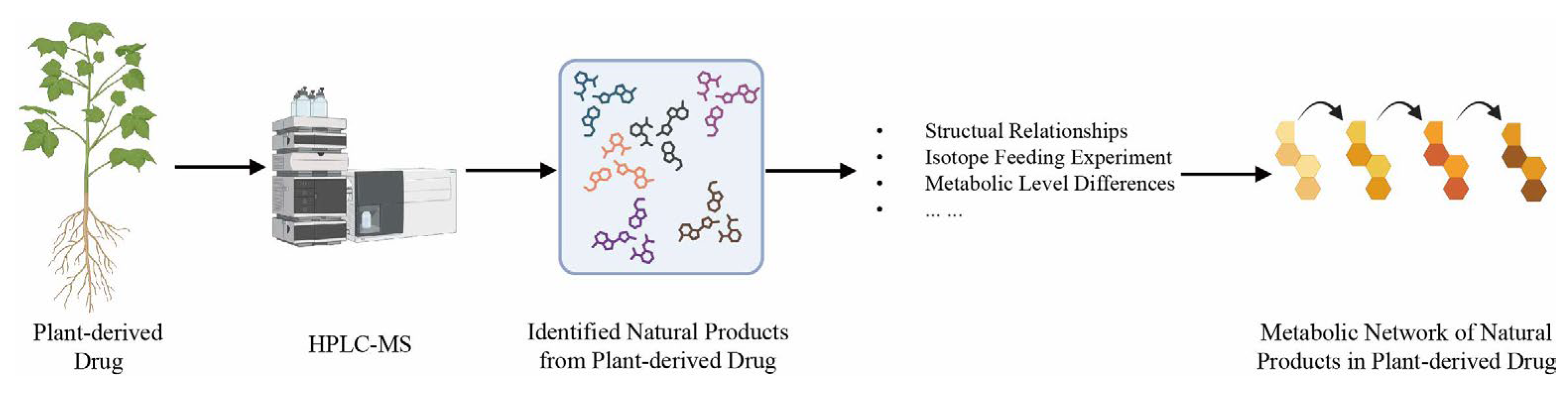
2.3. Pathway-Based Metabolic Network
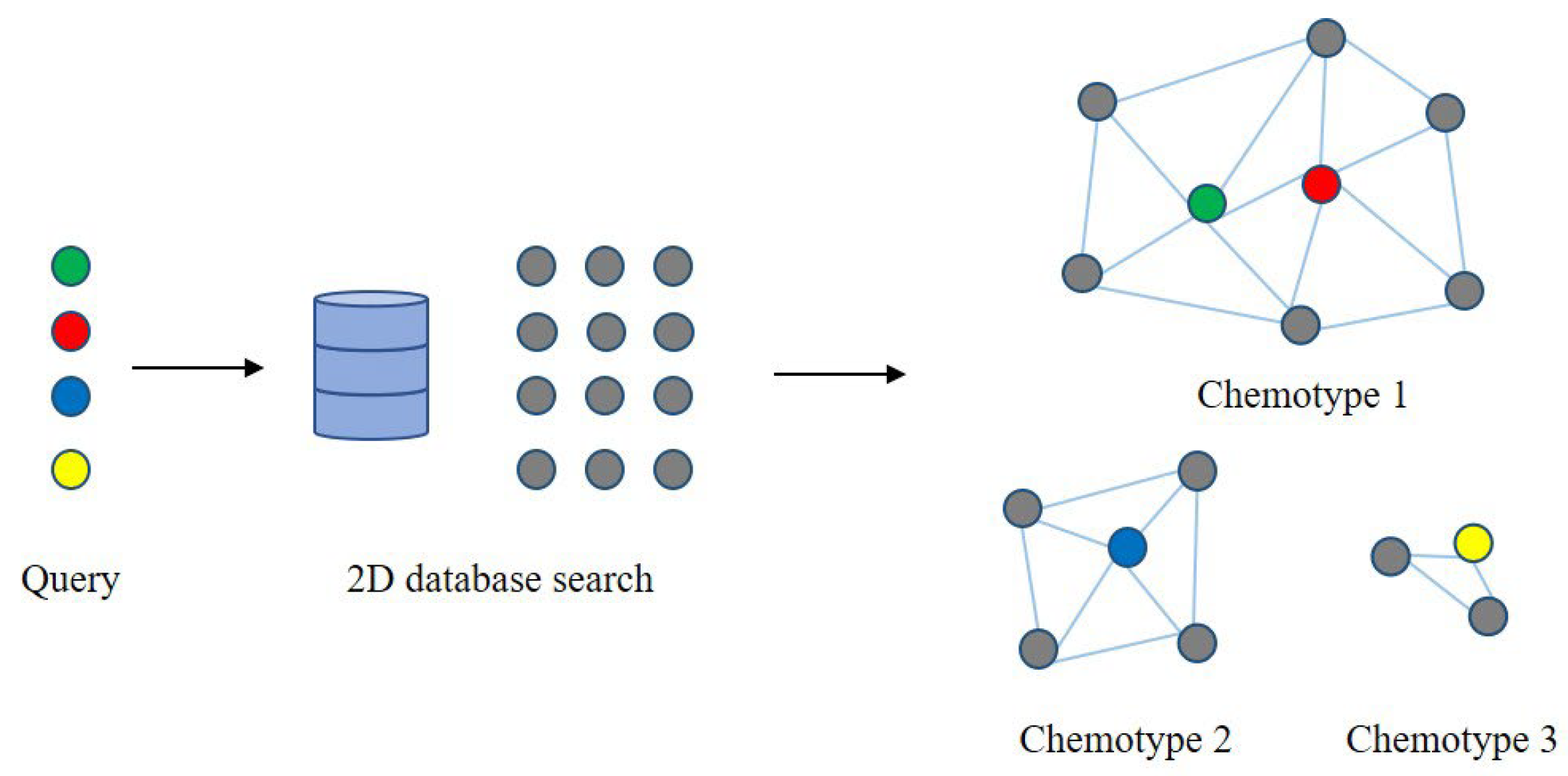
2.4. Metabolic Network Based on Chemical Structure Similarity
3. Application of Metabolic Network
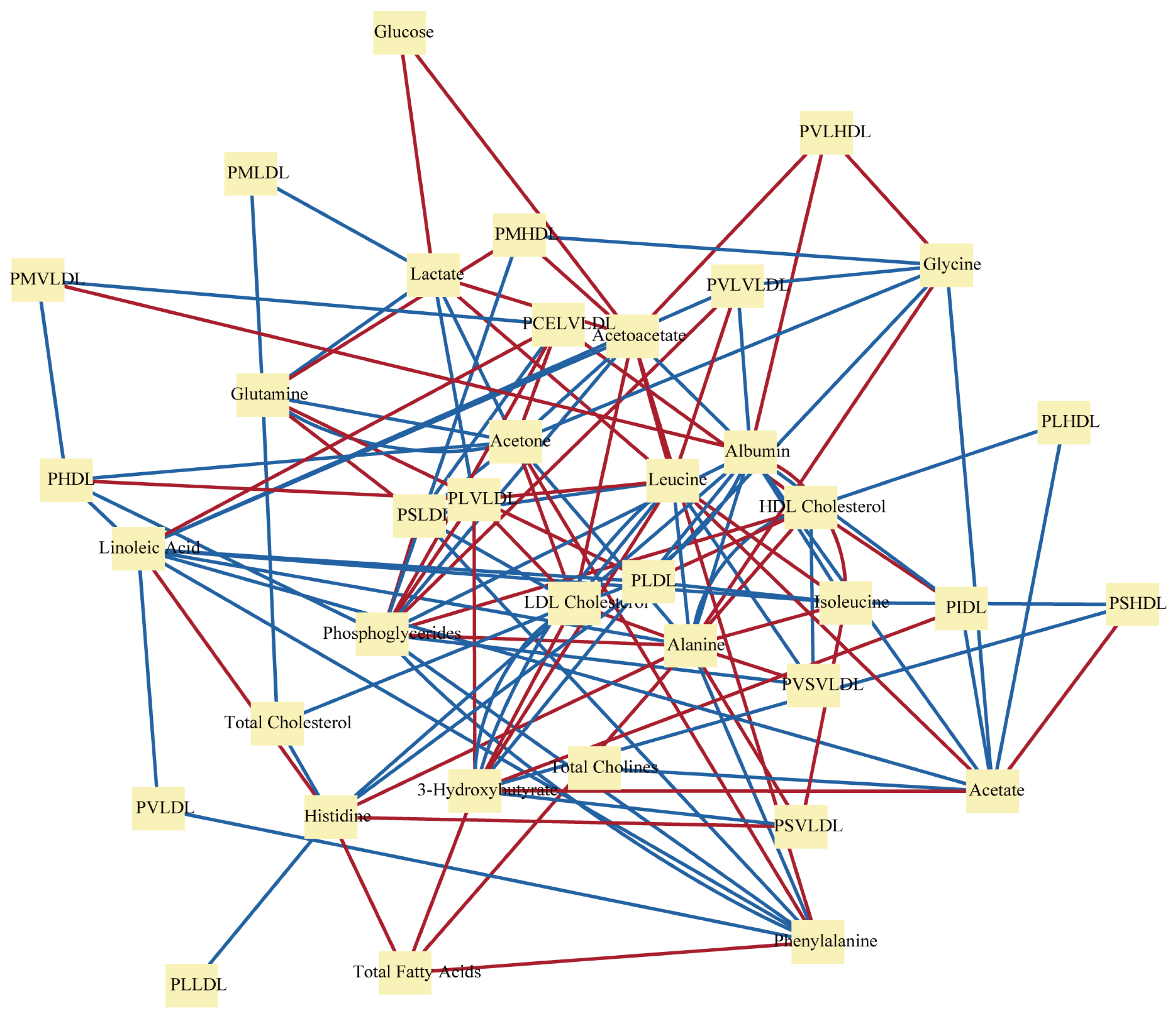
3.1. Metabolic Networks in Disease Mechanisms
3.2. Metabolic Networks in Disease Prediction and Diagnosis
3.3. Drug Discovery and Disease Treatment
4. Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Assenov, Y.; Ramirez, F.; Schelhorn, S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Muzio, G.; O’Bray, L.; Borgwardt, K. Biological network analysis with deep learning. Brief. Bioinform. 2021, 22, 1515–1530. [Google Scholar] [CrossRef]
- Klein, C.; Marino, A.; Sagot, M.F.; Vieira Milreu, P.; Brilli, M. Structural and dynamical analysis of biological networks. Brief. Funct. Genom. 2012, 11, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Mengiste, S.A.; Aertsen, A.; Kumar, A. Relevance of network topology for the dynamics of biological neuronal networks. bioRxiv 2021. [Google Scholar] [CrossRef]
- May, R.M. Network structure and the biology of populations. Trends Ecol. Evol. 2006, 21, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Melo, D.; Porto, A.; Cheverud, J.M.; Marroig, G. Modularity: Genes, development and evolution. Annu. Rev. Ecol. Evol. Syst. 2016, 47, 463–486. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, D.M.; Jeng, A.; Deem, M.W. The emergence of modularity in biological systems. Phys. Life Rev. 2011, 8, 129–160. [Google Scholar] [CrossRef] [PubMed]
- Lee, U.; Mashour, G.A. Role of Network Science in the Study of Anesthetic State Transitions. Anesthesiology 2018, 129, 1029–1044. [Google Scholar] [CrossRef]
- De Las Rivas, J.; Fontanillo, C. Protein-protein interactions essentials: Key concepts to building and analyzing interactome networks. PLoS Comput. Biol. 2010, 6, e1000807. [Google Scholar] [CrossRef]
- Wu, M.; Su, R.Q.; Li, X.; Ellis, T.; Lai, Y.C.; Wang, X. Engineering of regulated stochastic cell fate determination. Proc. Natl. Acad. Sci. USA 2013, 110, 10610–10615. [Google Scholar] [CrossRef]
- Folger, O.; Jerby, L.; Frezza, C.; Gottlieb, E.; Ruppin, E.; Shlomi, T. Predicting selective drug targets in cancer through metabolic networks. Mol. Syst. Biol. 2011, 7, 501. [Google Scholar] [CrossRef]
- Park, H.J.; Friston, K. Structural and functional brain networks: From connections to cognition. Science 2013, 342, 1238411. [Google Scholar] [CrossRef]
- Girvan, M.; Newman, M.E. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA 2002, 99, 7821–7826. [Google Scholar] [CrossRef]
- Amara, A.; Frainay, C.; Jourdan, F.; Naake, T.; Neumann, S.; Novoa-Del-Toro, E.M.; Salek, R.M.; Salzer, L.; Scharfenberg, S.; Witting, M. Networks and Graphs Discovery in Metabolomics Data Analysis and Interpretation. Front. Mol. Biosci. 2022, 9, 841373. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, A.; Yazdani, A.; Mendez-Giraldez, R.; Samiei, A.; Kosorok, M.R.; Schaid, D.J. From classical mendelian randomization to causal networks for systematic integration of multi-omics. Front. Genet. 2022, 13, 990486. [Google Scholar] [CrossRef] [PubMed]
- Ness, R.O.; Sachs, K.; Vitek, O. From Correlation to Causality: Statistical Approaches to Learning Regulatory Relationships in Large-Scale Biomolecular Investigations. J. Proteome Res. 2016, 15, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, J.M. Thinking Clearly About Correlations and Causation: Graphical Causal Models for Observational Data. Adv. Methods Pract. Psychol. Sci. 2018, 1, 27–42. [Google Scholar] [CrossRef]
- Hattori, M.; Okuno, Y.; Goto, S.; Kanehisa, M. Development of a chemical structure comparison method for integrated analysis of chemical and genomic information in the metabolic pathways. J. Am. Chem. Soc. 2003, 125, 11853–11865. [Google Scholar] [CrossRef] [PubMed]
- Holliday, G.L.; Andreini, C.; Fischer, J.D.; Rahman, S.A.; Almonacid, D.E.; Williams, S.T.; Pearson, W.R. MACiE: Exploring the diversity of biochemical reactions. Nucleic Acids Res. 2012, 40, D783–D789. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Carreño, C.; Torrecilla, J.L. dcor: Distance correlation and energy statistics in Python. SoftwareX 2023, 22, 101326. [Google Scholar] [CrossRef]
- Shimoni, Y.; Karavani, E.; Ravid, S.; Bak, P.; Ng, T.H.M.; Alford, S.H.; Meade, D.; Goldschmidt, Y. An Evaluation Toolkit to Guide Model Selection and Cohort Definition in Causal Inference. arXiv 2019, arXiv:1906.00442. [Google Scholar]
- Batushansky, A.; Toubiana, D.; Fait, A. Correlation-Based Network Generation, Visualization, and Analysis as a Powerful Tool in Biological Studies: A Case Study in Cancer Cell Metabolism. Biomed. Res. Int. 2016, 2016, 8313272. [Google Scholar] [CrossRef]
- Nishihara, R.; Glass, K.; Mima, K.; Hamada, T.; Nowak, J.A.; Qian, Z.R.; Kraft, P.; Giovannucci, E.L.; Fuchs, C.S.; Chan, A.T.; et al. Biomarker correlation network in colorectal carcinoma by tumor anatomic location. BMC Bioinform. 2017, 18, 304. [Google Scholar] [CrossRef]
- Kotze, H.L.; Armitage, E.G.; Sharkey, K.J.; Allwood, J.W.; Dunn, W.B.; Williams, K.J.; Goodacre, R. A novel untargeted metabolomics correlation-based network analysis incorporating human metabolic reconstructions. BMC Syst. Biol. 2013, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Cripps, A.W.; West, N.P.; Cox, A.J.; Zhang, P. A correlation-based network for biomarker discovery in obesity with metabolic syndrome. BMC Bioinform. 2019, 20, 477. [Google Scholar] [CrossRef]
- Jahagirdar, S.; Suarez-Diez, M.; Saccenti, E. Simulation and Reconstruction of Metabolite-Metabolite Association Networks Using a Metabolic Dynamic Model and Correlation Based Algorithms. J. Proteome Res. 2019, 18, 1099–1113. [Google Scholar] [CrossRef] [PubMed]
- de Siqueira Santos, S.; Takahashi, D.Y.; Nakata, A.; Fujita, A. A comparative study of statistical methods used to identify dependencies between gene expression signals. Brief. Bioinform. 2014, 15, 906–918. [Google Scholar] [CrossRef]
- Aguilera-Mendoza, L.; Marrero-Ponce, Y.; Garcia-Jacas, C.R.; Chavez, E.; Beltran, J.A.; Guillen-Ramirez, H.A.; Brizuela, C.A. Automatic construction of molecular similarity networks for visual graph mining in chemical space of bioactive peptides: An unsupervised learning approach. Sci. Rep. 2020, 10, 18074. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Nie, J.; Chen, H.S.; Ma, H.; Stewart, R.; Li, X.; Lu, M.Z.; Taylor, W.M.; Wei, H. Evaluation of gene association methods for coexpression network construction and biological knowledge discovery. PLoS ONE 2012, 7, e50411. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Moing, A.; Ebbels, T.M.; Maucourt, M.; Tomos, A.D.; Rolin, D.; Hooks, M.A. Correlation Network Analysis reveals a sequential reorganization of metabolic and transcriptional states during germination and gene-metabolite relationships in developing seedlings of Arabidopsis. BMC Syst. Biol. 2010, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Dyrba, M.; Mohammadi, R.; Grothe, M.J.; Kirste, T.; Teipel, S.J. Gaussian Graphical Models Reveal Inter-Modal and Inter-Regional Conditional Dependencies of Brain Alterations in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 99. [Google Scholar] [CrossRef]
- Krumsiek, J.; Suhre, K.; Illig, T.; Adamski, J.; Theis, F.J. Gaussian graphical modeling reconstructs pathway reactions from high-throughput metabolomics data. BMC Syst. Biol. 2011, 5, 21. [Google Scholar] [CrossRef]
- Hackett, S.R.; Baltz, E.A.; Coram, M.; Wranik, B.J.; Kim, G.; Baker, A.; Fan, M.; Hendrickson, D.G.; Berndl, M.; McIsaac, R.S. Learning causal networks using inducible transcription factors and transcriptome-wide time series. Mol. Syst. Biol. 2020, 16, e9174. [Google Scholar] [CrossRef] [PubMed]
- Perfetto, L.; Briganti, L.; Calderone, A.; Perpetuini, A.C.; Lannuccelli, M.; Langone, F.; Licata, L.; Marinkovic, M.; Mattioni, A.; Pavlidou, T.; et al. SIGNOR: A database of causal relationships between biological entities. Nucleic Acids Res. 2016, 44, D548–D554. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.Y.; Chu, Z.X.; Li, S.; Li, Y.L.; Gao, J.; Zhang, A.D. A Survey on Causal Inference. ACM Trans. Knowl. Discov. Data 2021, 15, 74. [Google Scholar] [CrossRef]
- Nogueira, A.R.; Pugnana, A.; Ruggieri, S.; Pedreschi, D.; Gama, J. Methods and tools for causal discovery and causal inference. Wiley Interdiscip. Rev.-Data Min. Knowl. Discov. 2022, 12, e1449. [Google Scholar] [CrossRef]
- Chang, R.; Trushina, E.; Zhu, K.; Zaidi, S.S.A.; Lau, B.M.; Kueider-Paisley, A.; Moein, S.; He, Q.; Alamprese, M.L.; Vagnerova, B.; et al. Predictive metabolic networks reveal sex- and APOE genotype-specific metabolic signatures and drivers for precision medicine in Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2023, 19, 518–531. [Google Scholar] [CrossRef]
- Glymour, C.; Zhang, K.; Spirtes, P. Review of Causal Discovery Methods Based on Graphical Models. Front. Genet. 2019, 10, 524. [Google Scholar] [CrossRef]
- Rosa, G.J.; Valente, B.D.; de los Campos, G.; Wu, X.L.; Gianola, D.; Silva, M.A. Inferring causal phenotype networks using structural equation models. Genet. Sel. Evol. 2011, 43, 6. [Google Scholar] [CrossRef]
- Friston, K. Dynamic causal modeling and Granger causality Comments on: The identification of interacting networks in the brain using fMRI: Model selection, causality and deconvolution. Neuroimage 2011, 58, 303–305, author reply 310–311. [Google Scholar] [CrossRef]
- Peters, J.; Janzing, D.; Schölkopf, B. Causal inference on time series using restricted structural equation models. Adv. Neural Inf. Process. Syst. 2013, 26, 154–162. [Google Scholar] [CrossRef]
- Rose, N.; Wagner, W.; Mayer, A.; Nagengast, B. Model-Based Manifest and Latent Composite Scores in Structural Equation Models. Collabra Psychol. 2019, 5, 9. [Google Scholar] [CrossRef]
- Bollen, K.A.; Hoyle, R.H. Latent variables in structural equation modeling. In Handbook of Structural Equation Modeling; Hoyle, R.H., Ed.; Guilford Press: New York, NY, USA, 2012; pp. 56–67. [Google Scholar]
- Yuan, K.-H.; Kouros, C.D.; Kelley, K. Diagnosis for covariance structure models by analyzing the path. Struct. Equ. Model. A Multidiscip. J. 2008, 15, 564–602. [Google Scholar] [CrossRef]
- Friston, K.J. Functional and effective connectivity: A review. Brain Connect. 2011, 1, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Rosato, A.; Tenori, L.; Cascante, M.; De Atauri Carulla, P.R.; Martins Dos Santos, V.A.P.; Saccenti, E. From correlation to causation: Analysis of metabolomics data using systems biology approaches. Metabolomics 2018, 14, 37. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, Y.; Teng, S.; Li, K. Prediction of sepsis mortality using metabolite biomarkers in the blood: A meta-analysis of death-related pathways and prospective validation. BMC Med. 2020, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, D471–D480. [Google Scholar] [CrossRef]
- King, Z.A.; Lu, J.; Drager, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsson, B.O.; Lewis, N.E. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2016, 44, D515–D522. [Google Scholar] [CrossRef]
- Henry, C.S.; DeJongh, M.; Best, A.A.; Frybarger, P.M.; Linsay, B.; Stevens, R.L. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 2010, 28, 977–982. [Google Scholar] [CrossRef]
- Wang, L.; Dash, S.; Ng, C.Y.; Maranas, C.D. A review of computational tools for design and reconstruction of metabolic pathways. Synth. Syst. Biotechnol. 2017, 2, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Hatzimanikatis, V.; Li, C.; Ionita, J.A.; Henry, C.S.; Jankowski, M.D.; Broadbelt, L.J. Exploring the diversity of complex metabolic networks. Bioinformatics 2005, 21, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-H.; Chang, W.-C.; Chiu, C.-M.; Huang, C.-C.; Huang, H.-D. FMM: A web server for metabolic pathway reconstruction and comparative analysis. Nucleic Acids Res. 2009, 37, W129–W134. [Google Scholar] [CrossRef] [PubMed]
- Pey, J.; Prada, J.; Beasley, J.E.; Planes, F.J. Path finding methods accounting for stoichiometry in metabolic networks. Genome Biol. 2011, 12, R49. [Google Scholar] [CrossRef]
- Delépine, B.; Duigou, T.; Carbonell, P.; Faulon, J.-L. RetroPath2. 0: A retrosynthesis workflow for metabolic engineers. Metab. Eng. 2018, 45, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.-C.; Torres, J.Z. Chemical similarity networks for drug discovery. Spec. Top. Drug Discov. 2016, 1, 53–70. [Google Scholar]
- Wale, N.; Watson, I.A.; Karypis, G. Comparison of descriptor spaces for chemical compound retrieval and classification. Knowl. Inf. Syst. 2008, 14, 347–375. [Google Scholar] [CrossRef]
- Hu, G.; Kuang, G.; Xiao, W.; Li, W.; Liu, G.; Tang, Y. Performance evaluation of 2D fingerprint and 3D shape similarity methods in virtual screening. J. Chem. Inf. Model. 2012, 52, 1103–1113. [Google Scholar] [CrossRef]
- Willett, P. Similarity searching using 2D structural fingerprints. Methods Mol. Biol. 2011, 672, 133–158. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminformatics 2015, 7, 20. [Google Scholar] [CrossRef]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Willett, P. Similarity-based data mining in files of two-dimensional chemical structures using fingerprint measures of molecular resemblance. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 2011, 1, 241–251. [Google Scholar] [CrossRef]
- Stumpfe, D.; Bajorath, J. Similarity searching. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 260–282. [Google Scholar] [CrossRef]
- Schmidt, R.; Ehmki, E.S.R.; Ohm, F.; Ehrlich, H.C.; Mashychev, A.; Rarey, M. Comparing Molecular Patterns Using the Example of SMARTS: Theory and Algorithms. J. Chem. Inf. Model. 2019, 59, 2560–2571. [Google Scholar] [CrossRef] [PubMed]
- Jeliazkova, N.; Kochev, N. AMBIT-SMARTS: Efficient Searching of Chemical Structures and Fragments. Mol. Inf. 2011, 30, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Goryanin, I. Human metabolic network reconstruction and its impact on drug discovery and development. Drug Discov. Today 2008, 13, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Bertrand, K.; Naviaux, J.C.; Monk, J.M.; Wells, A.; Wang, L.; Lingampelly, S.S.; Naviaux, R.K.; Chambers, C. Metabolomic and exposomic biomarkers of risk of future neurodevelopmental delay in human milk. Pediatr. Res. 2023, 93, 1710–1720. [Google Scholar] [CrossRef]
- Judge, A.; Dodd, M.S. Metabolism. Essays Biochem. 2020, 64, 607–647. [Google Scholar] [CrossRef]
- Dräger, A.; Planatscher, H. Metabolic Networks. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.-H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013; pp. 1249–1251. [Google Scholar]
- Waller, T.C.; Berg, J.A.; Lex, A.; Chapman, B.E.; Rutter, J. Compartment and hub definitions tune metabolic networks for metabolomic interpretations. GigaScience 2020, 9, giz137. [Google Scholar] [CrossRef]
- Theorell, A.; Stelling, J. Metabolic networks, microbial consortia, and analogies to smart grids. Proc. IEEE 2022, 110, 541–556. [Google Scholar] [CrossRef]
- Burke, P.E.P.; Campos, C.B.L.; Costa, L.D.F.; Quiles, M.G. A biochemical network modeling of a whole-cell. Sci. Rep. 2020, 10, 13303. [Google Scholar] [CrossRef]
- Frainay, C.; Jourdan, F. Computational methods to identify metabolic sub-networks based on metabolomic profiles. Brief. Bioinform. 2017, 18, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Agren, R.; Bordel, S.; Mardinoglu, A.; Pornputtapong, N.; Nookaew, I.; Nielsen, J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 2012, 8, e1002518. [Google Scholar] [CrossRef] [PubMed]
- Mehrmohamadi, M.; Liu, X.; Shestov, A.A.; Locasale, J.W. Characterization of the usage of the serine metabolic network in human cancer. Cell Rep. 2014, 9, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.R.; Preston, G.M.; Mithani, A. MAPPS: A Web-Based Tool for Metabolic Pathway Prediction and Network Analysis in the Postgenomic Era. ACS Synth. Biol. 2020, 9, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Väremo, L.; Nookaew, I.; Nielsen, J. Novel insights into obesity and diabetes through genome-scale metabolic modeling. Front. Physiol. 2013, 4, 92. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Croes, D.; van Helden, J. Prediction of metabolic pathways from genome-scale metabolic networks. Biosystems 2011, 105, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Hecht, F. On the origins of cancer genetics and cytogenetics. Cancer Genet. Cytogenet. 1987, 29, 187–190. [Google Scholar] [CrossRef]
- Bintener, T.; Pacheco, M.P.; Sauter, T. Towards the routine use of in silico screenings for drug discovery using metabolic modelling. Biochem. Soc. Trans. 2020, 48, 955–969. [Google Scholar] [CrossRef]
- Beguerisse-Díaz, M.; Bosque, G.; Oyarzún, D.; Picó, J.; Barahona, M. Flux-dependent graphs for metabolic networks. NPJ Syst. Biol. Appl. 2018, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Tomar, N.; De, R.K. Comparing methods for metabolic network analysis and an application to metabolic engineering. Gene 2013, 521, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zelezniak, A.; Pers, T.H.; Soares, S.; Patti, M.E.; Patil, K.R. Metabolic network topology reveals transcriptional regulatory signatures of type 2 diabetes. PLoS Comput. Biol. 2010, 6, e1000729. [Google Scholar] [CrossRef] [PubMed]
- Zimmet, P.; Alberti, K.G.; Shaw, J. Global and societal implications of the diabetes epidemic. Nature 2001, 414, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Hameed, I.; Masoodi, S.R.; Mir, S.A.; Nabi, M.; Ghazanfar, K.; Ganai, B.A. Type 2 diabetes mellitus: From a metabolic disorder to an inflammatory condition. World J. Diabetes 2015, 6, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhou, Y.; Tang, H.; Liu, B.; Su, B.; Wang, Q. Differential metabolic network construction for personalized medicine: Study of type 2 diabetes mellitus patients’ response to gliclazide-modified-release-treated. J. Biomed. Inform. 2021, 118, 103796. [Google Scholar] [CrossRef]
- Huang, X.; Wang, Z.; Su, B.; He, X.; Liu, B.; Kang, B. A computational strategy for metabolic network construction based on the overlapping ratio: Study of patients’ metabolic responses to different dialysis patterns. Comput. Biol. Chem. 2021, 93, 107539. [Google Scholar] [CrossRef]
- Hadjicharalambous, M.; Wijeratne, P.A.; Vavourakis, V. From tumour perfusion to drug delivery and clinical translation of in silico cancer models. Methods 2021, 185, 82–93. [Google Scholar] [CrossRef]
- Lee, M.H.; Hwang, Y.H.; Yun, C.S.; Han, B.S.; Kim, D.Y. Altered small-world property of a dynamic metabolic network in murine left hippocampus after exposure to acute stress. Sci. Rep. 2022, 12, 3885. [Google Scholar] [CrossRef]
- Sen, P.; Orešič, M. Integrating Omics Data in Genome-Scale Metabolic Modeling: A Methodological Perspective for Precision Medicine. Metabolites 2023, 13, 855. [Google Scholar] [CrossRef]
- Garcia-Segura, M.E.; Durainayagam, B.R.; Liggi, S.; Graça, G.; Jimenez, B.; Dehghan, A.; Tzoulaki, I.; Karaman, I.; Elliott, P.; Griffin, J.L. Pathway-based integration of multi-omics data reveals lipidomics alterations validated in an Alzheimer’s disease mouse model and risk loci carriers. J. Neurochem. 2023, 164, 57–76. [Google Scholar] [CrossRef] [PubMed]
- Bordbar, A.; Palsson, B.O. Using the reconstructed genome-scale human metabolic network to study physiology and pathology. J. Intern. Med. 2012, 271, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Capel, F.; Klimcáková, E.; Viguerie, N.; Roussel, B.; Vítková, M.; Kováciková, M.; Polák, J.; Kovácová, Z.; Galitzky, J.; Maoret, J.J.; et al. Macrophages and adipocytes in human obesity: Adipose tissue gene expression and insulin sensitivity during calorie restriction and weight stabilization. Diabetes 2009, 58, 1558–1567. [Google Scholar] [CrossRef] [PubMed]
- Fouladiha, H.; Marashi, S.A. Biomedical applications of cell- and tissue-specific metabolic network models. J. Biomed. Inform. 2017, 68, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Stempler, S.; Yizhak, K.; Ruppin, E. Integrating transcriptomics with metabolic modeling predicts biomarkers and drug targets for Alzheimer’s disease. PLoS ONE 2014, 9, e105383. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Berggren, J.R.; Hulver, M.W.; Houmard, J.A.; Hoffman, E.P. GRB14, GPD1, and GDF8 as potential network collaborators in weight loss-induced improvements in insulin action in human skeletal muscle. Physiol. Genom. 2006, 27, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.J.; Ma, W.; Li, Y.; Su, Y. Disease biomarker identification based on sample network optimization. Methods 2023, 213, 42–49. [Google Scholar] [CrossRef]
- Hu, Y.; Zhao, T.; Zhang, N.; Zang, T.; Zhang, J.; Cheng, L. Identifying diseases-related metabolites using random walk. BMC Bioinform. 2018, 19, 116. [Google Scholar] [CrossRef]
- Lei, X.; Tie, J. Prediction of disease-related metabolites using bi-random walks. PLoS ONE 2019, 14, e0225380. [Google Scholar] [CrossRef]
- Baumgartner, C.; Spath-Blass, V.; Niederkofler, V.; Bergmoser, K.; Langthaler, S.; Lassnig, A.; Rienmüller, T.; Baumgartner, D.; Asnani, A.; Gerszten, R.E. A novel network-based approach for discovering dynamic metabolic biomarkers in cardiovascular disease. PLoS ONE 2018, 13, e0208953. [Google Scholar] [CrossRef]
- Kerk, S.A.; Papagiannakopoulos, T.; Shah, Y.M.; Lyssiotis, C.A. Metabolic networks in mutant KRAS-driven tumours: Tissue specificities and the microenvironment. Nat. Rev. Cancer 2021, 21, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Hiller, K.; Metallo, C.M. Profiling metabolic networks to study cancer metabolism. Curr. Opin. Biotechnol. 2013, 24, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Charlestin, V.; Wang, M.; Walker, Z.T.; Miranda-Vergara, M.C.; Facchine, B.A.; Wu, J.; Kaliney, W.J.; Dovichi, N.J.; Li, J.; et al. Aquaporin-7 Regulates the Response to Cellular Stress in Breast Cancer. Cancer Res. 2020, 80, 4071–4086. [Google Scholar] [CrossRef] [PubMed]
- Bidkhori, G.; Benfeitas, R.; Klevstig, M.; Zhang, C.; Nielsen, J.; Uhlen, M.; Boren, J.; Mardinoglu, A. Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes. Proc. Natl. Acad. Sci. USA 2018, 115, E11874–E11883. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Zhou, G.; Ewald, J.; Chang, L.; Hacariz, O.; Basu, N.; Xia, J. Using MetaboAnalyst 5.0 for LC-HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat. Protoc. 2022, 17, 1735–1761. [Google Scholar] [CrossRef] [PubMed]
- Cicek, A.E.; Qi, X.; Cakmak, A.; Johnson, S.R.; Han, X.; Alshalwi, S.; Ozsoyoglu, Z.M.; Ozsoyoglu, G. An online system for metabolic network analysis. Database J. Biol. Databases Curation 2014, 2014, bau091. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ma, M.; Shen, N.; Xi, J.J.; Tian, W. Integration of cancer gene co-expression network and metabolic network to uncover potential cancer drug targets. J. Proteome Res. 2013, 12, 2354–2364. [Google Scholar] [CrossRef]
- Bidkhori, G.; Benfeitas, R.; Elmas, E.; Kararoudi, M.N.; Arif, M.; Uhlen, M.; Nielsen, J.; Mardinoglu, A. Metabolic Network-Based Identification and Prioritization of Anticancer Targets Based on Expression Data in Hepatocellular Carcinoma. Front. Physiol. 2018, 9, 916. [Google Scholar] [CrossRef]
- Kell, D.B.; Goodacre, R. Metabolomics and systems pharmacology: Why and how to model the human metabolic network for drug discovery. Drug Discov. Today 2014, 19, 171–182. [Google Scholar] [CrossRef]
- Shaked, I.; Oberhardt, M.A.; Atias, N.; Sharan, R.; Ruppin, E. Metabolic Network Prediction of Drug Side Effects. Cell Syst. 2016, 2, 209–213. [Google Scholar] [CrossRef]
- Chang, R.L.; Xie, L.; Xie, L.; Bourne, P.E.; Palsson, B. Drug off-target effects predicted using structural analysis in the context of a metabolic network model. PLoS Comput. Biol. 2010, 6, e1000938. [Google Scholar] [CrossRef]
- Qing, Z.; Yan, F.; Huang, P.; Zeng, J. Establishing the metabolic network of isoquinoline alkaloids from the Macleaya genus. Phytochemistry 2021, 185, 112696. [Google Scholar] [CrossRef] [PubMed]
- Karta, J.; Bossicard, Y.; Kotzamanis, K.; Dolznig, H.; Letellier, E. Mapping the Metabolic Networks of Tumor Cells and Cancer-Associated Fibroblasts. Cells 2021, 10, 304. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.E.; Abdel-Haleem, A.M. The evolution of genome-scale models of cancer metabolism. Front. Physiol. 2013, 4, 237. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, M.; Colombo, R.; Damiani, C.; Pescini, D.; Gaglio, D.; Vanoni, M.; Alberghina, L.; Mauri, G. Zooming-in on cancer metabolic rewiring with tissue specific constraint-based models. Comput. Biol. Chem. 2016, 62, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Kanhaiya, K.; Tyagi-Tiwari, D. Identification of Drug Targets in Breast Cancer Metabolic Network. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2020, 27, 975–986. [Google Scholar] [CrossRef] [PubMed]
- López-López, N.; León, D.S.; de Castro, S.; Díez-Martínez, R.; Iglesias-Bexiga, M.; Camarasa, M.J.; Menéndez, M.; Nogales, J.; Garmendia, J. Interrogation of Essentiality in the Reconstructed Haemophilus influenzae Metabolic Network Identifies Lipid Metabolism Antimicrobial Targets: Preclinical Evaluation of a FabH β-Ketoacyl-ACP Synthase Inhibitor. mSystems 2022, 7, e0145921. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Vásquez, T.; Fondi, M.; Perrin, E. Understanding Antimicrobial Resistance Using Genome-Scale Metabolic Modeling. Antibiotics 2023, 12, 896. [Google Scholar] [CrossRef] [PubMed]
- Mienda, B.S.; Salihu, R.; Adamu, A.; Idris, S. Genome-scale metabolic models as platforms for identification of novel genes as antimicrobial drug targets. Future Microbiol. 2018, 13, 455–467. [Google Scholar] [CrossRef]
- Mienda, B.S. Genome-scale metabolic models as platforms for strain design and biological discovery. J. Biomol. Struct. Dyn. 2017, 35, 1863–1873. [Google Scholar] [CrossRef]
- Chavali, A.K.; D’Auria, K.M.; Hewlett, E.L.; Pearson, R.D.; Papin, J.A. A metabolic network approach for the identification and prioritization of antimicrobial drug targets. Trends Microbiol. 2012, 20, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Durmuş, S.; Çakır, T.; Özgür, A.; Guthke, R. A review on computational systems biology of pathogen-host interactions. Front. Microbiol. 2015, 6, 235. [Google Scholar] [CrossRef]
- Cesur, M.F.; Siraj, B.; Uddin, R.; Durmuş, S.; Çakır, T. Network-Based Metabolism-Centered Screening of Potential Drug Targets in Klebsiella pneumoniae at Genome Scale. Front. Cell. Infect. Microbiol. 2019, 9, 447. [Google Scholar] [CrossRef]
- Nazarshodeh, E.; Marashi, S.A.; Gharaghani, S. Structural systems pharmacology: A framework for integrating metabolic network and structure-based virtual screening for drug discovery against bacteria. PLoS ONE 2021, 16, e0261267. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.M.; Mills, G.B.; Ram, P.T. Cancer Systems Biology: A peek into the future of patient care? Nat. Rev. Clin. Oncol. 2014, 11, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Zangene, E.; Marashi, S.A.; Montazeri, H. SL-scan identifies synthetic lethal interactions in cancer using metabolic networks. Sci. Rep. 2023, 13, 15763. [Google Scholar] [CrossRef]
- Guo, W.F.; Zhang, S.W.; Shi, Q.Q.; Zhang, C.M.; Zeng, T.; Chen, L. A novel algorithm for finding optimal driver nodes to target control complex networks and its applications for drug targets identification. BMC Genom. 2018, 19, 924. [Google Scholar] [CrossRef]
- Li, Y.L.; Wu, J.J.; Ma, J.; Li, S.S.; Xue, X.; Wei, D.; Shan, C.L.; Hua, X.Y.; Zheng, M.X.; Xu, J.G. Alteration of the Individual Metabolic Network of the Brain Based on Jensen-Shannon Divergence Similarity Estimation in Elderly Patients With Type 2 Diabetes Mellitus. Diabetes 2022, 71, 894–905. [Google Scholar] [CrossRef]
- Zhou, Z.; Luo, M.; Zhang, H.; Yin, Y.; Cai, Y.; Zhu, Z.J. Metabolite annotation from knowns to unknowns through knowledge-guided multi-layer metabolic networking. Nat. Commun. 2022, 13, 6656. [Google Scholar] [CrossRef]
- Huang, Y.; Xie, Y.; Zhong, C.; Zhou, F. Finding branched pathways in metabolic network via atom group tracking. PLoS Comput. Biol. 2021, 17, e1008676. [Google Scholar] [CrossRef]
- Cakmak, A.; Celik, M.H. Personalized Metabolic Analysis of Diseases. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Shah, H.A.; Liu, J.; Yang, Z.; Zhang, X.; Feng, J. DeepRF: A deep learning method for predicting metabolic pathways in organisms based on annotated genomes. Comput. Biol. Med. 2022, 147, 105756. [Google Scholar] [CrossRef] [PubMed]
- Onogi, A. A Bayesian model for genomic prediction using metabolic networks. Bioinform. Adv. 2023, 3, vbad106. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Network | Method/Model | Language | Source |
|---|---|---|---|
| Correlation-based | Pearson correlation And Spearman rank correlation | Python | https://github.com/aishapectyo/Correlations-Pearson-Spearman (accessed on 28 November 2023) |
| Distance correlation [20] | Python | https://github.com/vnmabus/dcor (accessed on 28 November 2023) | |
| Gaussian graphical model | R | https://github.com/donaldRwilliams/BGGM (accessed on 28 November 2023) | |
| Causal-based | Causal inference model [21] | Python | https://github.com/BiomedSciAI/causallib (accessed on 28 November 2023) |
| Structural equation model | R | https://github.com/yrosseel/lavaan (accessed on 28 November 2023) | |
| Dynamic causal model | Python | https://github.com/tmdemelo/pydcm (accessed on 28 November 2023) | |
| Pathway-based | Pathway | Python | https://github.com/iseekwonderful/PyPathway (accessed on 28 November 2023) |
| Chemical structure similarity-based | Chemical structure similarity | Python | https://github.com/labsyspharm/lsp-cheminformatics (accessed on 28 November 2023) |
| No. | Tool | Application | Character | URL |
|---|---|---|---|---|
| 1 | MAPPS [78] | A web-based tool for pathway prediction and network comparison, identification of potential drug targets | Allow users to upload custom data. | https://mapps.lums.edu.pk (accessed on 30 November 2023) |
| 2 | MetaboAnalyst [107] | A Network Explorer module for integrative analysis of metabolomics, metagenomics, and/or transcriptomics data. | For comprehensive metabolomic data analysis, interpretation, and integration with other omics data. | https://metaboanalyst.ca/ (accessed on 30 November 2023) |
| 3 | PathCase [108] | A database-enabled framework and Web-based computational tools for browsing, querying, analyzing, and visualizing stored metabolic networks. | Create a new metabolic network and/or update an existing metabolic network. The network can also be created from an existing genome-scale reconstructed network. | http://nashua.case.edu/PathwaysMAW/Web (accessed on 30 November 2023) |
| 4 | Met-express [109] | A powerful tool for uncovering novel therapeutic biomarkers. | Integrate a cancer gene co-expression network with the metabolic network to predict key enzyme-coding genes and metabolites in cancer cell metabolism. | None |
| 5 | Baumgartner C et al. [102] | A novel network-based approach for discovering dynamic metabolic biomarkers in cardiovascular disease. | Combine metabolic time-series data into a superimposed graph representation, highlighting the strength of the underlying kinetic interaction of preselected analytes. | None |
| 6 | Bidkhori et al. [110] | A metabolic network-based tool for identification and prioritization of anticancer targets. | Predict and rank potential anticancer non-toxic controlling metabolite and gene targets. | None |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, W.; Pan, H.; Sha, Y.; Zhai, X.; Xing, A.; Lingampelly, S.S.; Sripathi, S.R.; Wang, Y.; Li, K. Metabolic Connectome and Its Role in the Prediction, Diagnosis, and Treatment of Complex Diseases. Metabolites 2024, 14, 93. https://doi.org/10.3390/metabo14020093
Meng W, Pan H, Sha Y, Zhai X, Xing A, Lingampelly SS, Sripathi SR, Wang Y, Li K. Metabolic Connectome and Its Role in the Prediction, Diagnosis, and Treatment of Complex Diseases. Metabolites. 2024; 14(2):93. https://doi.org/10.3390/metabo14020093
Chicago/Turabian StyleMeng, Weiyu, Hongxin Pan, Yuyang Sha, Xiaobing Zhai, Abao Xing, Sai Sachin Lingampelly, Srinivasa R. Sripathi, Yuefei Wang, and Kefeng Li. 2024. "Metabolic Connectome and Its Role in the Prediction, Diagnosis, and Treatment of Complex Diseases" Metabolites 14, no. 2: 93. https://doi.org/10.3390/metabo14020093
APA StyleMeng, W., Pan, H., Sha, Y., Zhai, X., Xing, A., Lingampelly, S. S., Sripathi, S. R., Wang, Y., & Li, K. (2024). Metabolic Connectome and Its Role in the Prediction, Diagnosis, and Treatment of Complex Diseases. Metabolites, 14(2), 93. https://doi.org/10.3390/metabo14020093