
Exploring the Impact of Organic Solvent Quality and Unusual Adduct Formation during LC-MS-Based Lipidomic Profiling

 ,
,  and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Sample Preparation
2.3. LC-MS Conditions
2.4. Data Processing
3. Results and Discussion
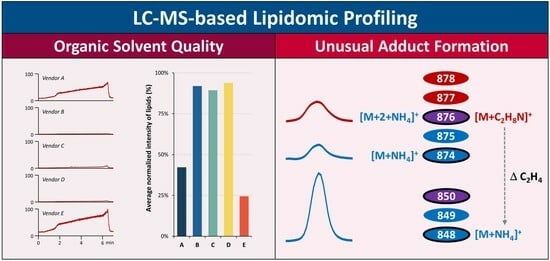
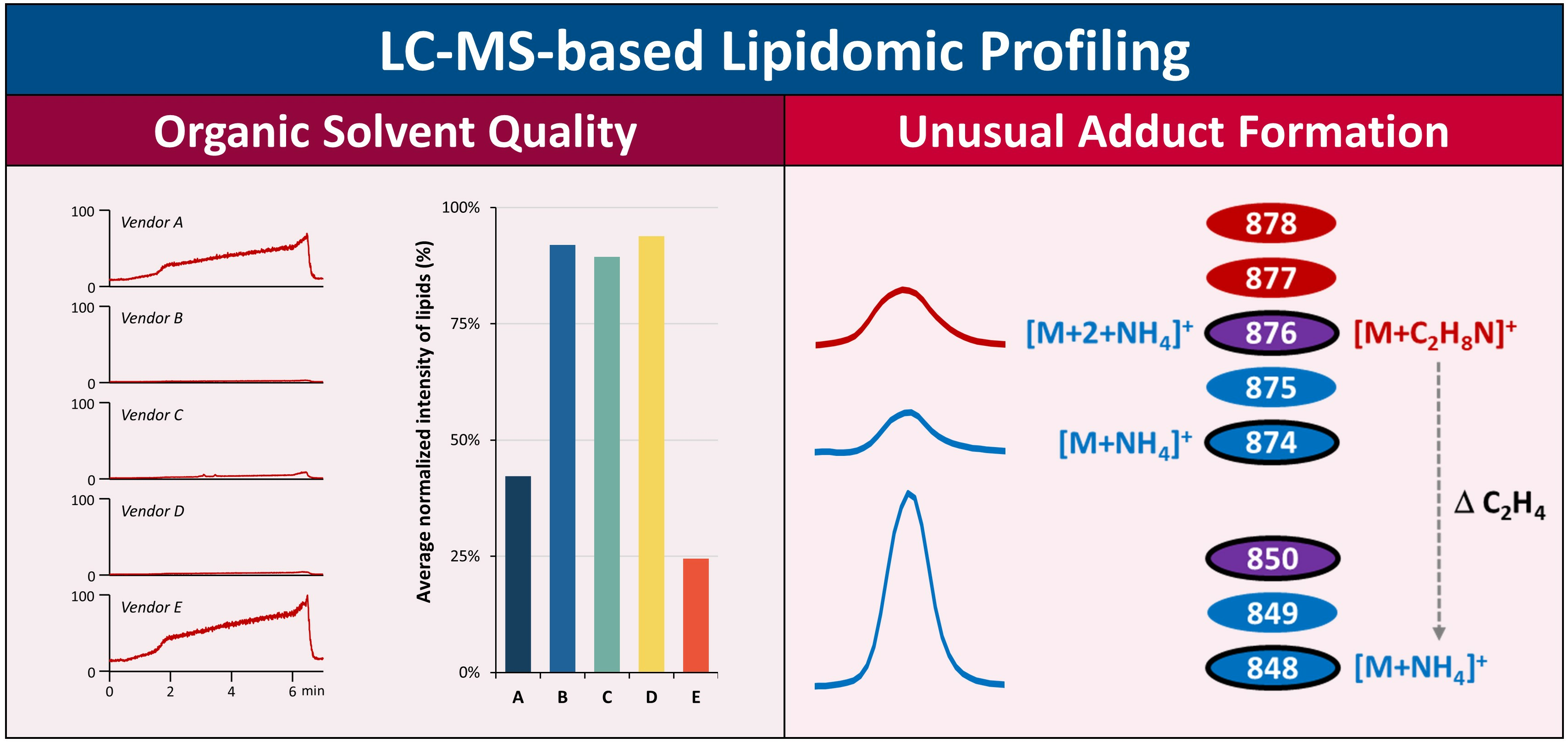
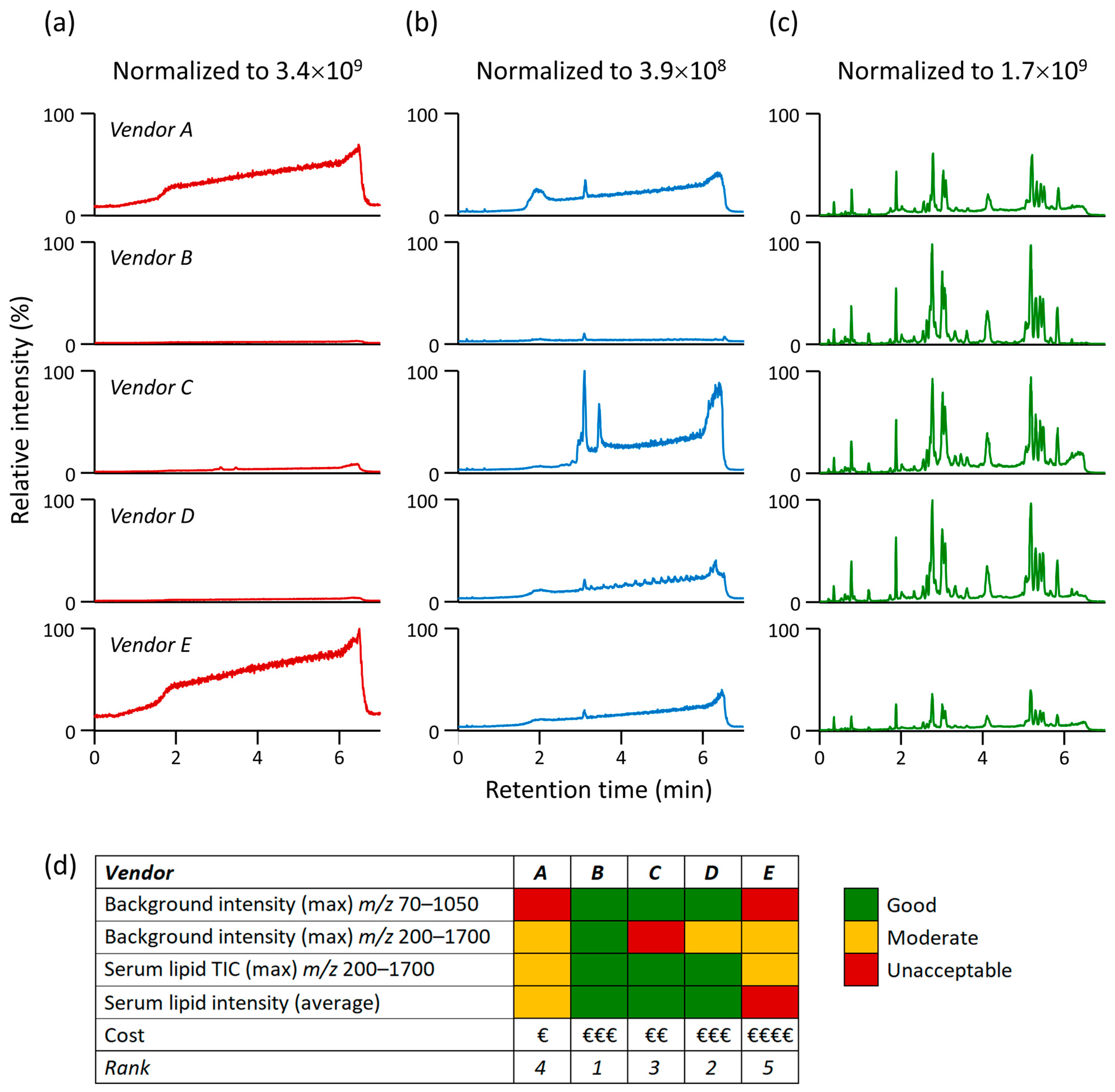
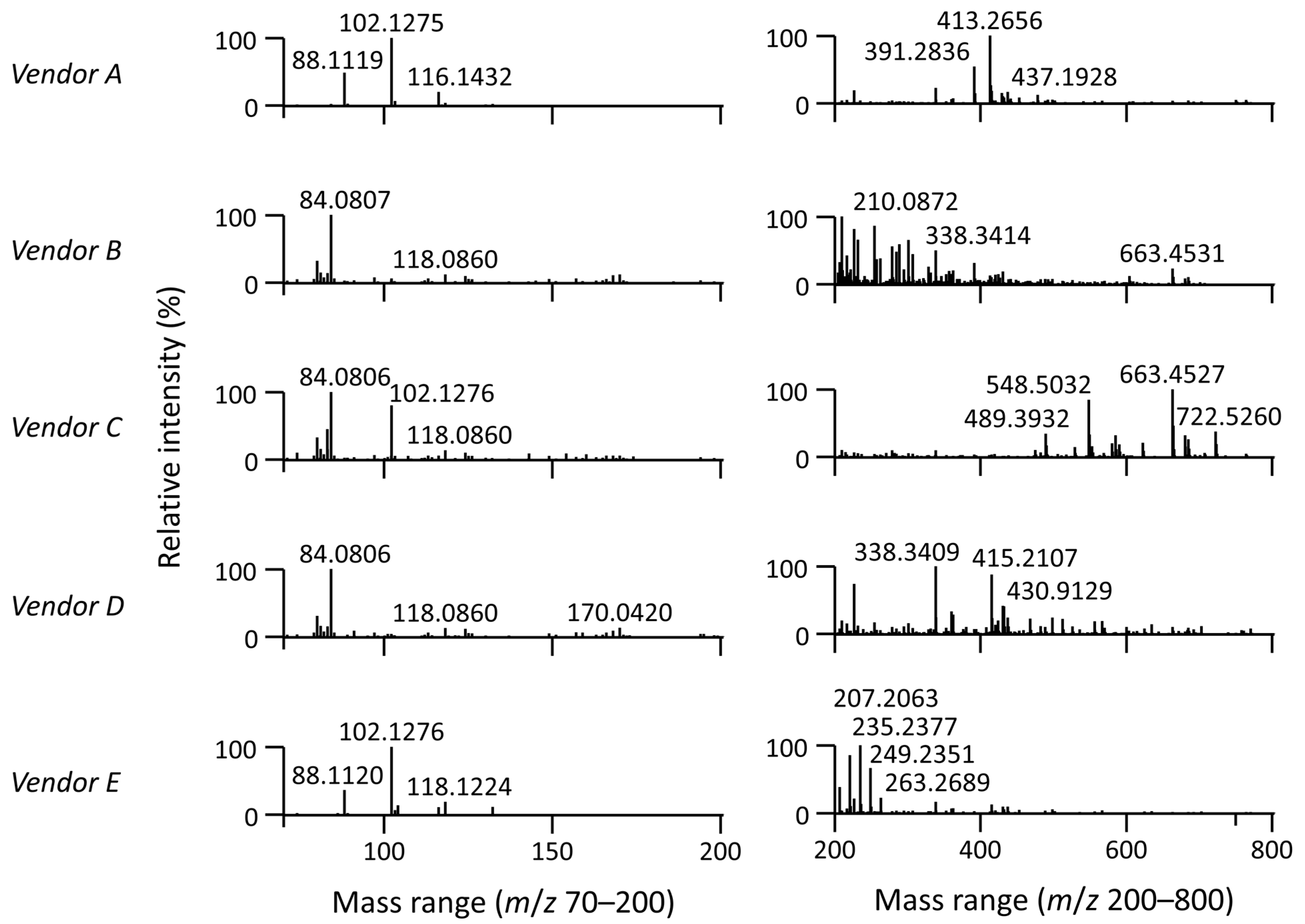
3.1. Impact of Organic Solvent Quality during LC-MS-Based Lipidomic Profiling
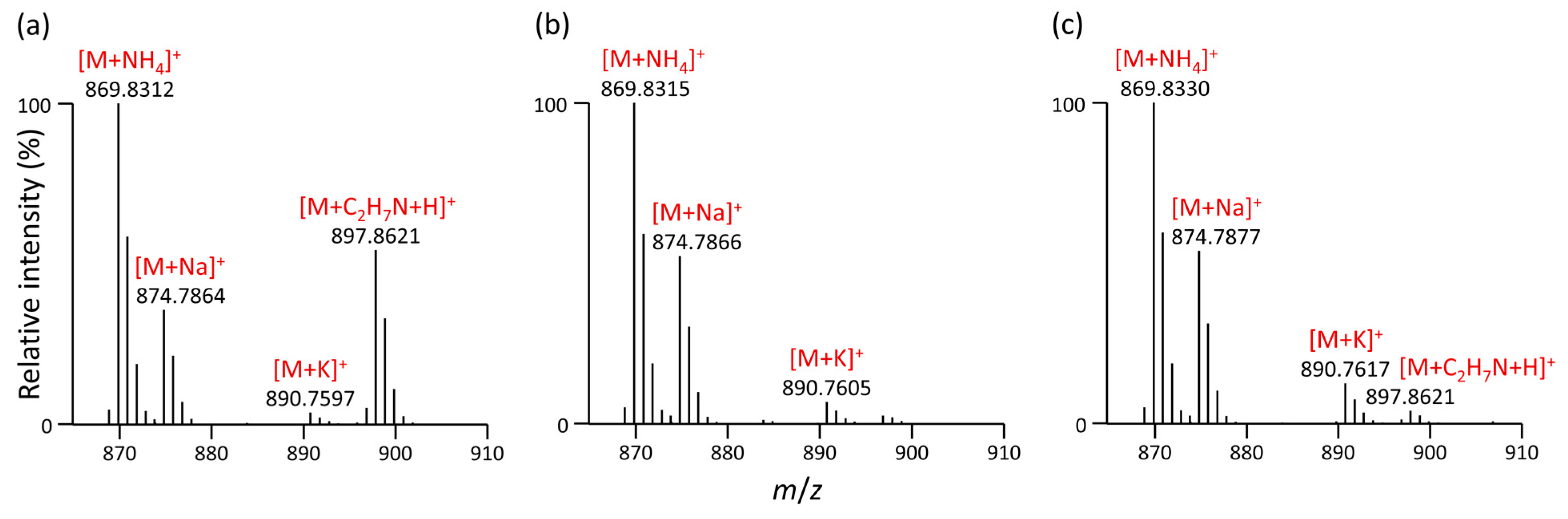
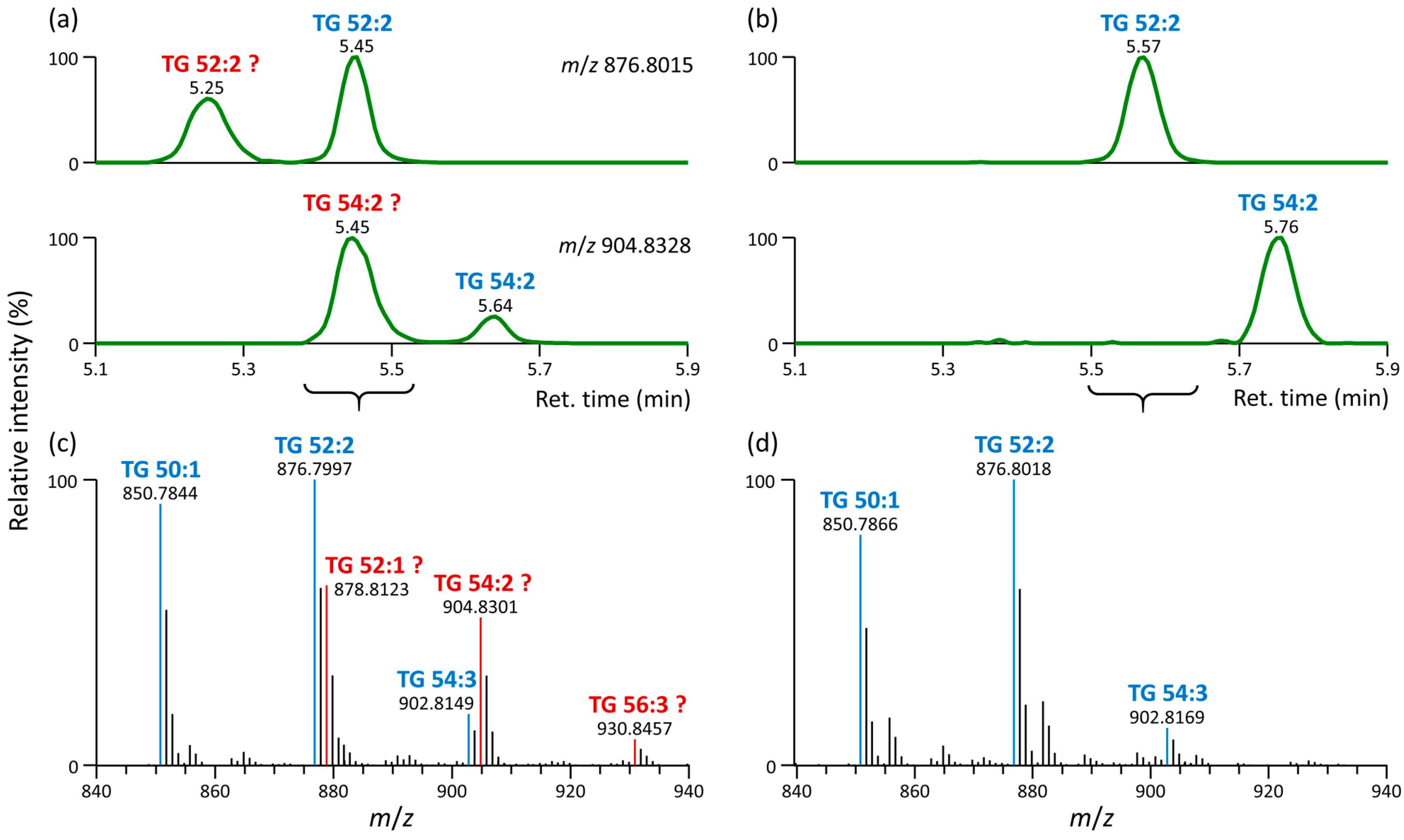
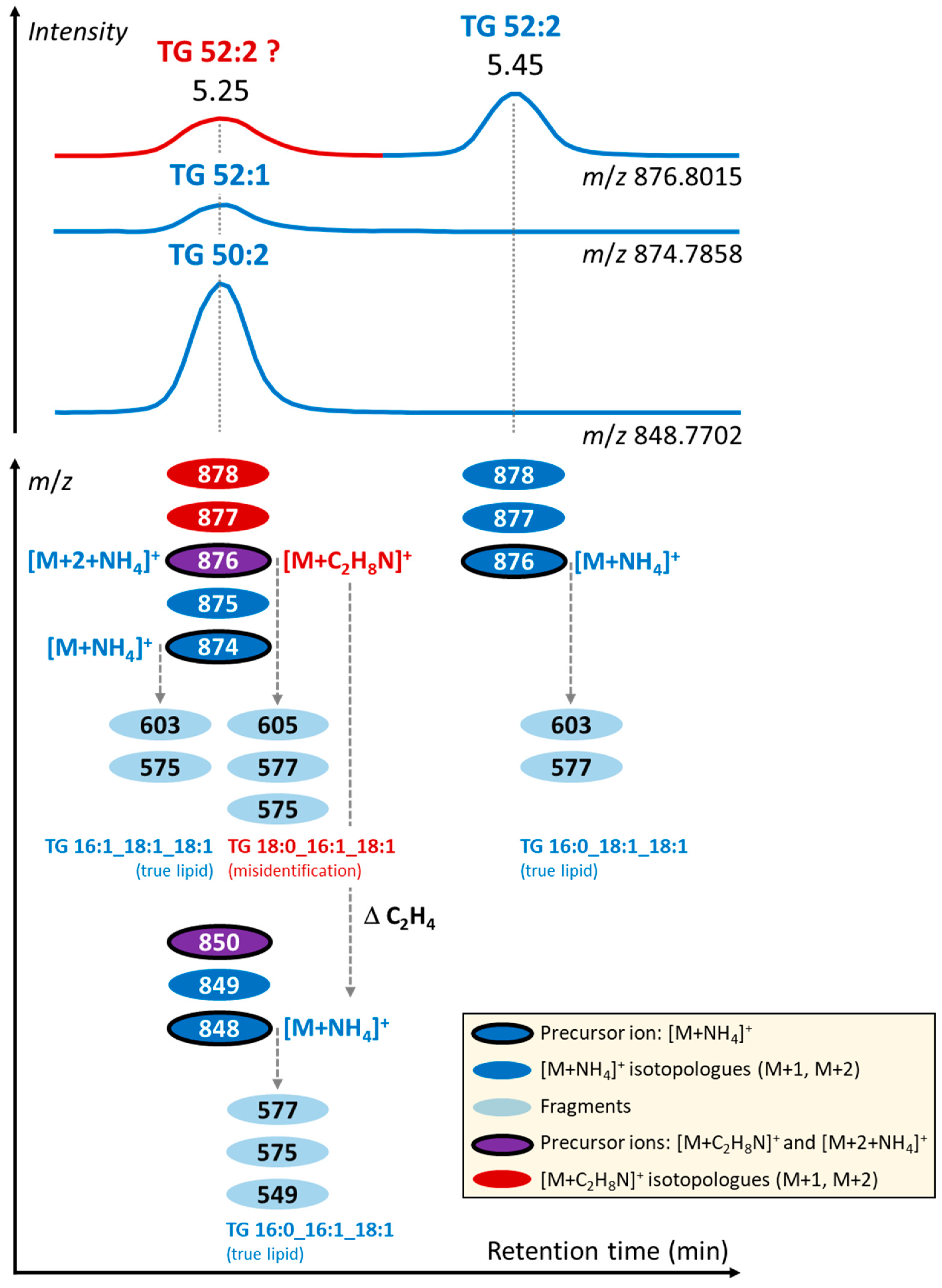
3.2. Formation of Unusual Adducts during LC-MS-Based Lipidomic Profiling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rustam, Y.H.; Reid, G.E. Analytical challenges and recent advances in mass spectrometry based lipidomics. Anal. Chem. 2018, 90, 374–397. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Fiehn, O. Toward merging untargeted and targeted methods in mass spectrometry-based metabolomics and lipidomics. Anal. Chem. 2016, 88, 524–545. [Google Scholar] [CrossRef] [PubMed]
- Rampler, E.; El Abiead, Y.; Schoeny, H.; Rusz, M.; Hildebrand, F.; Fitz, V.; Koellensperger, G. Recurrent topics in mass spectrometry-based metabolomics and lipidomics-standardization, coverage, and throughput. Anal. Chem. 2021, 93, 519–545. [Google Scholar] [CrossRef] [PubMed]
- Kostidis, S.; Sanchez-Lopez, E.; Giera, M. Lipidomics analysis in drug discovery and development. Curr. Opin. Chem. Biol. 2023, 72, 102256. [Google Scholar] [CrossRef]
- Géhin, C.; Fowler, S.J.; Trivedi, D.K. Chewing the fat: How lipidomics is changing our understanding of human health and disease in 2022. Anal. Sci. Adv. 2023, 4, 104–131. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. TrAC-Trend Anal. Chem. 2014, 61, 192–206. [Google Scholar] [CrossRef]
- Miller, K.E.; Jorgenson, J.W. Comparison of microcapillary column length and inner diameter investigated with gradient analysis of lipids by ultrahigh-pressure liquid chromatography-mass spectrometry. J. Sep. Sci. 2020, 43, 4094–4102. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Increasing lipidomic coverage by selecting optimal mobile-phase modifiers in LC–MS of blood plasma. Metabolomics 2016, 12, 34. [Google Scholar] [CrossRef]
- Narvaez-Rivas, M.; Zhang, Q.B. Comprehensive untargeted lipidomic analysis using core-shell C30 particle column and high field orbitrap mass spectrometer. J. Chromatogr. A 2016, 1440, 123–134. [Google Scholar] [CrossRef]
- Lange, M.; Fedorova, M. Evaluation of lipid quantification accuracy using HILIC and RPLC MS on the example of NIST (R) SRM (R) 1950 metabolites in human plasma. Anal. Bioanal. Chem. 2020, 412, 3573–3584. [Google Scholar] [CrossRef]
- Criscuolo, A.; Zeller, M.; Cook, K.; Angelidou, G.; Fedorova, M. Rational selection of reverse phase columns for high throughput LC-MS lipidomics. Chem. Phys. Lipids 2019, 221, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Contrepois, K.; Mahmoudi, S.; Ubhi, B.K.; Papsdorf, K.; Hornburg, D.; Brunet, A.; Snyder, M. Cross-platform comparison of untargeted and targeted lipidomics approaches on aging mouse plasma. Sci. Rep. 2018, 8, 17747. [Google Scholar] [CrossRef]
- Cajka, T.; Hricko, J.; Rudl Kulhava, L.; Paucova, M.; Novakova, M.; Kuda, O. Optimization of mobile phase modifiers for fast LC-MS-based untargeted metabolomics and lipidomics. Int. J. Mol. Sci. 2023, 24, 1987. [Google Scholar] [CrossRef] [PubMed]
- Monnin, C.; Ramrup, P.; Daigle-Young, C.; Vuckovic, D. Improving negative liquid chromatography/electrospray ionization mass spectrometry lipidomic analysis of human plasma using acetic acid as a mobile-phase additive. Rapid Commun. Mass. Sp. 2018, 32, 201–211. [Google Scholar] [CrossRef]
- Creydt, M.; Fischer, M. Plant metabolomics: Maximizing metabolome coverage by optimizing mobile phase additives for nontargeted mass spectrometry in positive and negative electrospray ionization mode. Anal. Chem. 2017, 89, 10474–10486. [Google Scholar] [CrossRef]
- Lenk, A. Solvents: An overlooked ally for liquid chromatography–mass spectrometry. Column 2018, 14, 19–21. [Google Scholar]
- Lupo, S. LC-MS sensitivity: Practical strategies to boost your signal and lower your noise. LC GC Eur. 2018, 31, 512–520. [Google Scholar]
- Ni, Z.X.; Wolk, M.; Jukes, G.; Espinosa, K.M.; Ahrends, R.; Aimo, L.; Alvarez-Jarreta, J.; Andrews, S.; Andrews, R.; Bridge, A.; et al. Guiding the choice of informatics software and tools for lipidomics research applications. Nat. Methods 2023, 20, 193–204. [Google Scholar] [CrossRef]
- Kind, T.; Liu, K.H.; Lee do, Y.; DeFelice, B.; Meissen, J.K.; Fiehn, O. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef]
- Kyle, J.E.; Crowell, K.L.; Casey, C.P.; Fujimoto, G.M.; Kim, S.; Dautel, S.E.; Smith, R.D.; Payne, S.H.; Metz, T.O. LIQUID: An-open source software for identifying lipids in LC-MS/MS-based lipidomics data. Bioinformatics 2017, 33, 1744–1746. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.X.; Angelidou, G.; Lange, M.; Hoffmann, R.; Fedorova, M. LipidHunter identifies phospholipids by high-throughput processing of LC-MS and shotgun lipidomics datasets. Anal. Chem. 2017, 89, 8800–8807. [Google Scholar] [CrossRef] [PubMed]
- Rakusanova, S.; Fiehn, O.; Cajka, T. Toward building mass spectrometry-based metabolomics and lipidomics atlases for biological and clinical research. TrAC-Trend Anal. Chem. 2023, 158, 116825. [Google Scholar] [CrossRef]
- Kofeler, H.C.; Eichmann, T.O.; Ahrends, R.; Bowden, J.A.; Danne-Rasche, N.; Dennis, E.A.; Fedorova, M.; Griffiths, W.J.; Han, X.L.; Hartler, J.; et al. Quality control requirements for the correct annotation of lipidomics data. Nat. Commun. 2021, 12, 4771. [Google Scholar] [CrossRef]
- Hricko, J.; Kulhava, L.R.; Paucova, M.; Novakova, M.; Kuda, O.; Fiehn, O.; Cajka, T. Short-term stability of serum and liver extracts for untargeted metabolomics and lipidomics. Antioxidants 2023, 12, 986. [Google Scholar] [CrossRef]
- Janovska, P.; Melenovsky, V.; Svobodova, M.; Havlenova, T.; Kratochvilova, H.; Haluzik, M.; Hoskova, E.; Pelikanova, T.; Kautzner, J.; Monzo, L.; et al. Dysregulation of epicardial adipose tissue in cachexia due to heart failure: The role of natriuretic peptides and cardiolipin. J. Cachexia Sarcopenia Muscle 2020, 11, 1614–1627. [Google Scholar] [CrossRef]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef]
- Kirwan, J.A.; Gika, H.; Beger, R.D.; Bearden, D.; Dunn, W.B.; Goodacre, R.; Theodoridis, G.; Witting, M.; Yu, L.R.; Wilson, I.D. Quality assurance and quality control reporting in untargeted metabolic phenotyping: mQACC recommendations for analytical quality management. Metabolomics 2022, 18, 70. [Google Scholar] [CrossRef]
- Benke, P.I.; Burla, B.; Ekroos, K.; Wenk, M.R.; Torta, F. Impact of ion suppression by sample cap liners in lipidomics. Anal. Chim. Acta. 2020, 1137, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Lippa, K.A.; Aristizabal-Henao, J.J.; Beger, R.D.; Bowden, J.A.; Broeckling, C.; Beecher, C.; Clay Davis, W.; Dunn, W.B.; Flores, R.; Goodacre, R.; et al. Reference materials for MS-based untargeted metabolomics and lipidomics: A review by the metabolomics quality assurance and quality control consortium (mQACC). Metabolomics 2022, 18, 24. [Google Scholar] [CrossRef] [PubMed]
- Kofeler, H.C.; Ahrends, R.; Baker, E.S.; Ekroos, K.; Han, X.L.; Hoffmann, N.; Holcapek, M.; Wenk, M.R.; Liebisch, G. Recommendations for good practice in MS-based lipidomics. J. Lipid Res. 2021, 62, 100138. [Google Scholar] [CrossRef]
- Page, J.S.; Bogdanov, B.; Vilkov, A.N.; Prior, D.C.; Buschbach, M.A.; Tang, K.; Smith, R.D. Automatic gain control in mass spectrometry using a jet disrupter electrode in an electrodynamic ion funnel. J. Am. Soc. Mass Spectr. 2005, 16, 244–253. [Google Scholar] [CrossRef][Green Version]
- Urban, M.; Enot, D.P.; Dallmann, G.; Korner, L.; Forcher, V.; Enoh, P.; Koal, T.; Keller, M.; Deigner, H.P. Complexity and pitfalls of mass spectrometry-based targeted metabolomics in brain research. Anal. Biochem. 2010, 406, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Keller, B.O.; Suj, J.; Young, A.B.; Whittal, R.M. Interferences and contaminants encountered in modern mass spectrometry. Anal. Chim. Acta 2008, 627, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Fernandez, J.; Puello-Polo, E.; Lopez-Martinez, J. Recovery of (Z)-13-docosenamide from industrial wastewater and its application in the production of virgin polypropylene to improve the coefficient of friction in film type applications. Sustainability 2023, 15, 1247. [Google Scholar] [CrossRef]
- Hermabessiere, L.; Receveur, J.; Himber, C.; Mazurais, D.; Huvet, A.; Lagarde, F.; Lambert, C.; Paul-Pont, I.; Dehaut, A.; Jezequel, R.; et al. An Irgafos (R) 168 story: When the ubiquity of an additive prevents studying its leaching from plastics. Sci. Total. Environ. 2020, 749, 141651. [Google Scholar] [CrossRef] [PubMed]
- Du, B.B.; Shen, M.J.; Pan, Z.B.; Zhu, C.Y.; Luo, D.; Zeng, L.X. Trace analysis of multiple synthetic phenolic antioxidants in foods by liquid chromatography-tandem mass spectrometry with complementary use of electrospray ionization and atmospheric pressure chemical ionization. Food Chem. 2022, 375, 131663. [Google Scholar] [CrossRef]
- Lange, M.; Angelidou, G.; Ni, Z.X.; Criscuolo, A.; Schiller, J.; Bluher, M.; Fedorova, M. AdipoAtlas: A reference lipidome for human white adipose tissue. Cell Rep. Med. 2021, 2, 100407. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.A.; Heckert, A.; Ulmer, C.Z.; Jones, C.M.; Koelmel, J.P.; Abdullah, L.; Ahonen, L.; Alnouti, Y.; Armando, A.M.; Asara, J.M.; et al. Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950-Metabolites in Frozen Human Plasma. J. Lipid Res. 2017, 58, 2275–2288. [Google Scholar] [CrossRef]
- Stoll, D.R. Contaminants everywhere! Tips and tricks for reducing background signals when using LC-MS. LC GC N. Am. 2018, 36, 498–504. [Google Scholar]
- Koelmel, J.P.; Li, X.D.; Stow, S.M.; Sartain, M.J.; Murali, A.; Kemperman, R.; Tsugawa, H.; Takahashi, M.; Vasiliou, V.; Bowden, J.A.; et al. Lipid Annotator: Towards accurate annotation in non-targeted liquid chromatography high-resolution tandem mass spectrometry (LC-HRMS/MS) lipidomics using a rapid and user-friendly software. Metabolites 2020, 10, 101. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. LC-MS-based lipidomics and automated identification of lipids using the LipidBlast in-silico MS/MS library. Methods Mol. Biol. 2017, 1609, 149–170. [Google Scholar] [CrossRef]
- Koelmel, J.P.; Ulmer, C.Z.; Jones, C.M.; Yost, R.A.; Bowden, J.A. Common cases of improper lipid annotation using high-resolution tandem mass spectrometry data and corresponding limitations in biological interpretation. BBA-Mol. Cell Biol. L. 2017, 1862, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Kruve, A.; Kaupmees, K. Adduct formation in ESI/MS by mobile phase additives. J. Am. Soc. Mass Spectr. 2017, 28, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Riano, C.; Gradillas, A.; Barbas, C. Exploiting the formation of adducts in mobile phases with ammonium fluoride for the enhancement of annotation in liquid chromatography-high resolution mass spectrometry based lipidomics. J. Chromatogr. A Open 2021, 1, 100018. [Google Scholar] [CrossRef]
- Gu, Z.M.; Ma, J.Y.; Zhao, X.G.; Wu, J.; Zhang, D.L. Reduction of nitriles to amines in positive ion electrospray ionization mass spectrometry. Rapid Commun. Mass Sp. 2006, 20, 2969–2972. [Google Scholar] [CrossRef]
- Licea-Perez, H.; Junnotula, V.; Zohrabian, S.; Karlinsey, M. Development of a multi-sugar LC-MS/MS assay using simple chemical derivatization with acetic anhydride. Anal. Methods 2016, 8, 3023–3033. [Google Scholar] [CrossRef]
- Byrdwell, W.C.; Kotapati, H.K.; Goldschmidt, R. Fast chromatography of pulse triacylglycerols. J. Am. Oil Chem. Soc. 2023, 100, 25–43. [Google Scholar] [CrossRef]
- Brejchova, K.; Radner, F.P.W.; Balas, L.; Paluchova, V.; Cajka, T.; Chodounska, H.; Kudova, E.; Schratter, M.; Schreiber, R.; Durand, T.; et al. Distinct roles of adipose triglyceride lipase and hormone-sensitive lipase in the catabolism of triacylglycerol estolides. Proc. Natl. Acad. Sci. USA 2021, 118, e2020999118. [Google Scholar] [CrossRef]
- Wang, Y.F.; Harrington, P.D.; Chen, P. Metabolomic profiling and comparison of major cinnamon species using UHPLC-HRMS. Anal. Bioanal. Chem. 2020, 412, 7669–7681. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cajka, T.; Hricko, J.; Rudl Kulhava, L.; Paucova, M.; Novakova, M.; Fiehn, O.; Kuda, O. Exploring the Impact of Organic Solvent Quality and Unusual Adduct Formation during LC-MS-Based Lipidomic Profiling. Metabolites 2023, 13, 966. https://doi.org/10.3390/metabo13090966
Cajka T, Hricko J, Rudl Kulhava L, Paucova M, Novakova M, Fiehn O, Kuda O. Exploring the Impact of Organic Solvent Quality and Unusual Adduct Formation during LC-MS-Based Lipidomic Profiling. Metabolites. 2023; 13(9):966. https://doi.org/10.3390/metabo13090966
Chicago/Turabian StyleCajka, Tomas, Jiri Hricko, Lucie Rudl Kulhava, Michaela Paucova, Michaela Novakova, Oliver Fiehn, and Ondrej Kuda. 2023. "Exploring the Impact of Organic Solvent Quality and Unusual Adduct Formation during LC-MS-Based Lipidomic Profiling" Metabolites 13, no. 9: 966. https://doi.org/10.3390/metabo13090966
APA StyleCajka, T., Hricko, J., Rudl Kulhava, L., Paucova, M., Novakova, M., Fiehn, O., & Kuda, O. (2023). Exploring the Impact of Organic Solvent Quality and Unusual Adduct Formation during LC-MS-Based Lipidomic Profiling. Metabolites, 13(9), 966. https://doi.org/10.3390/metabo13090966