



Novel Metabolomic Approach for Identifying Pathology-Specific Biomarkers in Rare Diseases: A Case Study in Oculopharyngeal Muscular Dystrophy (OPMD)

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of Murine Samples
2.2. Oculopharyngeal Muscular Dystrophy Patient Samples
2.3. Chemicals and Reagents
2.4. Preparation of Tissue and Plasma Samples
2.5. LC–MS Analysis of Aqueous Phase (HILIC)
2.6. LC–MS Analysis of Non-Aqueous Phase (Reversed Phase)
2.7. Data Processing and Statistical Analysis
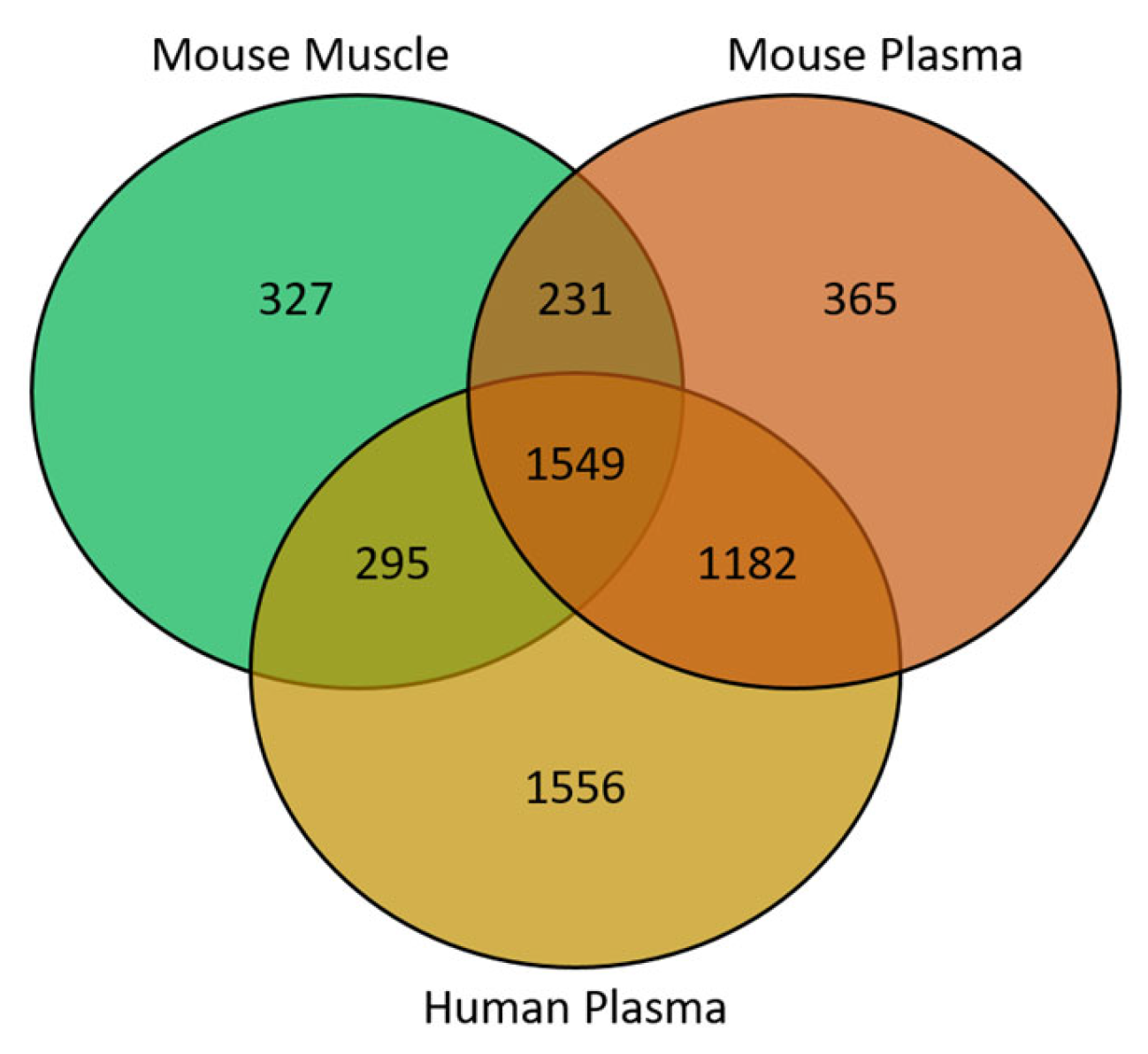
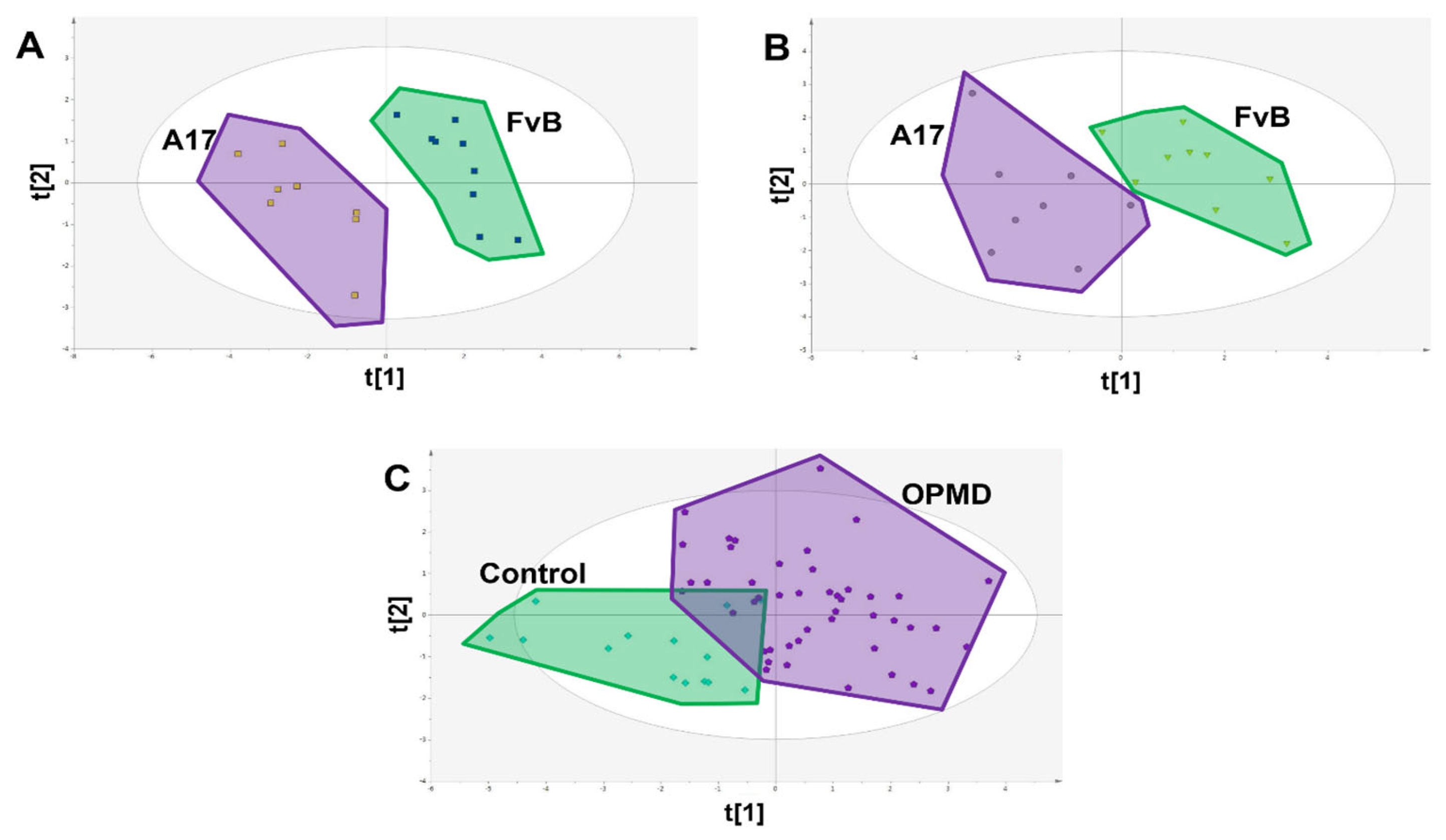
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organisation. International Programme on Chemical Safety Biomarkers in Risk Assessment: Validity and Validation; World Health Organisation: Geneva, Switzerland, 2001. [Google Scholar]
- Strimbu, K.; Tavel, J.A. What are Biomarkers? Curr. Opin. HIV AIDS 2010, 5, 463–466. [Google Scholar] [CrossRef]
- Ballman, K.V. Biomarker: Predictive or Prognostic? J. Clin. Oncol. 2015, 33, 3968–3971. [Google Scholar] [CrossRef]
- Kerr, D.J.; Yang, L. Personalising cancer medicine with prognostic markers. eBioMedicine 2021, 72, 103577. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Gulshan, K.; Nguyen, T.; Wu, Y. Biomarkers of Cardiovascular Disease. Dis. Markers 2017, 2017, 8208609. [Google Scholar] [CrossRef] [PubMed]
- Tolstikov, V.; Moser, A.J.; Sarangarajan, R.; Narain, N.R.; Kiebish, M.A. Current status of metabolomic biomarker discovery: Impact of study design and demographic characteristics. Metabolites 2020, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Pinu, F.R.; Beale, D.J.; Pate, A.M.; Kouremenos, K.; Swarup, S.; Schirra, H.J.; Wishart, D.W. Systems biology and mutli-omics integration: Viewpoints from the metabolomics research community. Metabolites 2019, 9, 76. [Google Scholar] [CrossRef]
- Hardikar, S.; Albrechtsen, R.D.; Achaintre, D.; Lin, T.; Pauleck, S.; Playdon, M.; Holowatyj, A.N.; Gigic, B.; Schrotz-King, P.; Boehm, J.; et al. Impact of pre-blood collection factors on plasma metabolomic profiles. Metabolites 2020, 10, 213. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.R.; Patel, R.; Kirsch, D.G.; Lewis, C.A.; Vander Heiden, M.G.; Locasale, J.W. Metabolomics in cancer research and emerging applications in clinical oncology. CA Cancer J. Clin. 2021, 71, 333–358. [Google Scholar] [CrossRef]
- Harshfield, E.L.; Fauman, E.B.; Stacey, D.; Paul, D.S.; Ziemek, D.; Ong, R.M.Y.; Danesh, J.; Butterworth, A.S.; Rasheed, A.; Sattar, T.; et al. Genome-wide analysis of blood lipid metabolites in over 5000 South Asians reveals biological insights at cardiometabolic disease loci. BMC Med. 2021, 19, 232. [Google Scholar] [CrossRef]
- Snowden, S.G.; Korosi, A.; de Rooij, S.R.; Koulman, A. Combining lipidomics and machine learning to measure clinical lipids in dried blood spots. Metabolomics 2020, 16, 83. [Google Scholar] [CrossRef]
- Abu-Baker, A.; Rouleau, G.A. Oculopharyngeal muscular dystrophy: Recent advances in the understanding of the molecular pathogenic mechanisms and treatment strategies. Biochim. Biophys. Acta–Mol. Basis Dis. 2007, 1772, 173–185. [Google Scholar] [CrossRef]
- Harish, P.; Malerba, A.; Dickson, G.; Bachtarzi, H. Progress on gene therapy, cell therapy, and pharmacological strategies toward the treatment of Oculopharyngeal Muscular Dystrophy. Hum. Gene Ther. 2015, 26, 286–292. [Google Scholar] [CrossRef]
- Grosse, S.D.; Rogowski, W.H.; Ross, L.F.; Cornel, M.C.; Dondrop, W.J.; Khoury, M.J. Populations screening for genetic disorders in the 21st century: Evidence, economics and ethics. Public Health Genom. 2010, 13, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Harish, P.; Malerba, A.; Lu-Nguyen, N.; Forrest, L.; Cappellari, O.; Roth, F.; Trollet, C.; Popplewell, L.; Dickson, G. Inhibition of myostatin improves muscle atrophy in oculopharyngeal muscular dystrophy (OPMD). J. Cachexia Sarcopenia Muscle 2019, 10, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Trollet, C.; Anvar, S.Y.; Venema, A.; Hargreaves, I.P.; Foster, K.; Vignaud, A.; Ferry, A.; Negroni, E.; Hourde, C.; Baraibar, M.A.; et al. Molecular and phenotypic characterization of a mouse model of oculopharyngeal muscular dystrophy reveals severe muscular atrophy restricted to fast glycolytic fibres. Hum. Mol. Genet. 2010, 19, 2191–2207. [Google Scholar] [CrossRef] [PubMed]
- Malerba, A.; Roth, F.; Harish, P.; Dhiab, J.; Lu-Nguyen, N.; Cappellari, O.; Jarmin, S.; Mahoudeau, A.; Ythier, V.; Laine, J.; et al. Pharmacological modulation of the ER stress response ameliorates oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 2019, 28, 1694–1708. [Google Scholar] [CrossRef] [PubMed]
- Malerba, A.; Klein, P.; Bachtarzi, H.; Jarmin, S.A.; Cordova, G.; Ferry, A.; Strings, V.; Polay Espinoza, M.; Mamchaoui, K.; Blumen, S.C.; et al. PABPN1 gene therapy for oculopharyngeal muscular dystrophy. Nat. Commun. 2017, 8, 14848. [Google Scholar] [CrossRef]
- Raz, V.; Kroon, R.H.M.J.M.; Mei, H.; Riaz, M.; Buermans, H.; Lassche, S.; Horlings, C.; De Swart, B.; Kalf, J.; Harish, P.; et al. Age-associated salivary microRNA biomarkers for oculopharyngeal muscular dystrophy. Int. J. Mol. Sci. 2020, 21, 6059. [Google Scholar] [CrossRef]
- de Leeuw, R.H.; Garnier, D.; Kroon, R.M.J.M.; Horlings, C.G.C.; de Meijer, E.; Buermans, H.; van Engelen, B.G.M.; de Knijff, P.; Raz, V. Diagnostics of short tandem repeat expansion variants using massively parallel sequencing and componential tools. Eur. J. Hum. Gen. 2019, 27, 400–407. [Google Scholar] [CrossRef]
- Ebshiana, A.A.; Snowden, S.G.; Thambisetty, M.; Parsons, R.; Hye, A.; Legido-Quigley, C. Metabolomics method: UPLC-q-ToF polar and non-polar metabolites in the healthy rat cerebellum using an in-vial dual extraction. PLoS ONE 2015, 10, e0122883. [Google Scholar] [CrossRef]
- Fernandes, H.J.R.; Kent, J.P.; Bruntraeger, M.; Bassett, A.R.; Koulman, A.; Metzakopian, E.; Snowden, S.G. Mitochondrial and endoplasmic reticulum stress trigger triglyceride accumulation in models of Parkinson’s disease independent of mutations in MAPT. Metabolites 2023, 13, 112. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, C.; Tautenhahn, R.; Bottcher, C.; Larson, T.R.; Neumann, S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry datasets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Eosu, K.; Young-Sang, J.; Hyunjeong, K.; Jin-Sup, K.; Minsun, P.; Jihyeon, J.; Ho-Geun, Y.; Geum-Sook, H.; Kee, N. Metabolomic signatures in peripheral blood associated with Alzheimer’s disease Amyloid-β-induced neuroinflammation. J. Alzheimers Dis. 2014, 42, 421–433. [Google Scholar]
- Moreau, R.; Claria, J.; Aguilar, F.; Fenaille, F.; Lozano, J.J.; Junot, C.; Colsch, B.; Caraceni, P.; Trebicka, J.; Pavesi, M.; et al. Blood metabolomics uncovers inflammation-associated mitochondrial dysfunction as a potential mechanism underlying ACLF. J. Hepatol. 2020, 72, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, S.D.; Mukherjee, S.; Sharp, S.J.; Proitsi, P.; Lotta, L.A.; Day, F.; Perry, J.R.B.; Boehme, K.L.; Walter, S.; Kauwe, J.S.; et al. Association between potentially modifiable risk factors and Alzheimer’s Disease: A mendelian randomization study. PLoS Med. 2015, 12, e1001841. [Google Scholar] [CrossRef]
- Davies, N.M.; Holmes, M.V.; Smith, G.D. Reading mendelian randomisation studies: A guide, glossary and checklist for clinicians. BMJ 2018, 362, k601. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | OPMD | |
|---|---|---|
| Sample N° (% female) | 14 (50%) | 51 (55%) |
| Age (years) | 61.0 ± 12.8 | 60.4 ± 8.5 |
| Mean age of onset (years) | n/a | 50.5 ± 7.4 |
| Muscle 1 | Muscle 2 | Mouse Plasma | Human Plasma | |||||
|---|---|---|---|---|---|---|---|---|
| p-Value | FC+ | p-Value | FC+ | p-Value | FC+ | p-Value | FC++ | |
| HILIC_4 | 0.035 | 0.81 | 0.033 | 0.78 | 0.018 | 0.64 | 0.0041 | 0.62 |
| HILIC_14 | 8.22 × 10−5 | 0.76 | 0.022 | 0.38 | 0.021 | 0.76 | 1.56 × 10−5 | 0.71 |
| HILIC_55 | 8.22 × 10−5 | 1.43 | 0.0006 | 2.07 | 0.040 | 0.82 | 0.0055 | 0.89 |
| HILIC_186 | 0.0079 | 0.79 | 0.037 | 0.71 | 0.094 | 0.76 | 0.027 | 0.79 |
| HILIC_201 | 0.0079 | 0.88 | 0.026 | 0.70 | 0.034 | 0.84 | 0.0073 | 0.84 |
| RP_1362 | 0.015 | 0.79 | 0.0029 | 0.65 | 0.079 | 0.80 | 0.0023 | 0.72 |
| HILIC_687 | 0.029 | 0.63 | 0.020 | 0.513 | 0.027 | 1.26 | 0.013 | 5.88 |
| HILIC_2523 | 0.020 | 0.83 | 0.031 | 0.49 | 0.028 | 1.30 | 0.038 | 1.39 |
| RP_1362 | 0.0005 | 0.44 | 0.0024 | 0.49 | 0.069 | 1.32 | 0.0073 | 1.74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harish, P.; Malerba, A.; Kroon, R.H.M.J.M.; Shademan, M.; van Engelan, B.; Raz, V.; Popplewell, L.; Snowden, S.G. Novel Metabolomic Approach for Identifying Pathology-Specific Biomarkers in Rare Diseases: A Case Study in Oculopharyngeal Muscular Dystrophy (OPMD). Metabolites 2023, 13, 769. https://doi.org/10.3390/metabo13060769
Harish P, Malerba A, Kroon RHMJM, Shademan M, van Engelan B, Raz V, Popplewell L, Snowden SG. Novel Metabolomic Approach for Identifying Pathology-Specific Biomarkers in Rare Diseases: A Case Study in Oculopharyngeal Muscular Dystrophy (OPMD). Metabolites. 2023; 13(6):769. https://doi.org/10.3390/metabo13060769
Chicago/Turabian StyleHarish, Pradeep, Alberto Malerba, Rosemarie H. M. J. M. Kroon, Milad Shademan, Baziel van Engelan, Vered Raz, Linda Popplewell, and Stuart G. Snowden. 2023. "Novel Metabolomic Approach for Identifying Pathology-Specific Biomarkers in Rare Diseases: A Case Study in Oculopharyngeal Muscular Dystrophy (OPMD)" Metabolites 13, no. 6: 769. https://doi.org/10.3390/metabo13060769
APA StyleHarish, P., Malerba, A., Kroon, R. H. M. J. M., Shademan, M., van Engelan, B., Raz, V., Popplewell, L., & Snowden, S. G. (2023). Novel Metabolomic Approach for Identifying Pathology-Specific Biomarkers in Rare Diseases: A Case Study in Oculopharyngeal Muscular Dystrophy (OPMD). Metabolites, 13(6), 769. https://doi.org/10.3390/metabo13060769