



Changes in the Secretion of Melatonin and Selected Adipokines during the Progression of Parkinson’s Disease—Preliminary Studies

 ,
,  ,
,  , , and
, , and 
Abstract
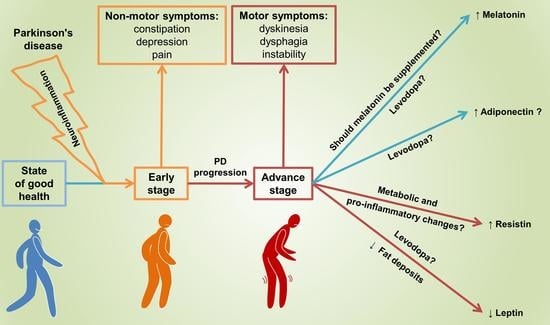
1. Introduction
2. Material and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Armstrong, M.J.; Okun, M.S. Time for a New Image of Parkinson Disease. JAMA Neurol. 2020, 77, 1345. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease. JAMA 2020, 323, 548. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s Disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Gustafson, B.; Jack, M.M.; Cushman, S.W.; Smith, U. Adiponectin Gene Activation by Thiazolidinediones Requires PPARγ2, but Not C/EBPα—Evidence for Differential Regulation of the AP2 and Adiponectin Genes. Biochem. Biophys. Res. Commun. 2003, 308, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. Alpha-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef]
- Salari, M.; Barzegar, M.; Etemadifar, M.; Mirmosayyeb, O. Serum Leptin Levels in Iranian Patients with Parkinson’s Disease. Iran. J. Neurol. 2018, 17, 71–77. [Google Scholar]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Daneshvar Kakhaki, R.; Ostadmohammadi, V.; Kouchaki, E.; Aghadavod, E.; Bahmani, F.; Tamtaji, O.R.; Reiter, R.J.; Mansournia, M.A.; Asemi, Z. Melatonin Supplementation and the Effects on Clinical and Metabolic Status in Parkinson’s Disease: A Randomized, Double-Blind, Placebo-Controlled Trial. Clin. Neurol. Neurosurg. 2020, 195, 105878. [Google Scholar] [CrossRef]
- Ahn, J.H.; Kim, M.; Park, S.; Jang, W.; Park, J.; Oh, E.; Cho, J.W.; Kim, J.S.; Youn, J. Prolonged-Release Melatonin in Parkinson’s Disease Patients with a Poor Sleep Quality: A Randomized Trial. Parkinsonism Relat. Disord. 2020, 75, 50–54. [Google Scholar] [CrossRef]
- Hadi, F.; Agah, E.; Tavanbakhsh, S.; Mirsepassi, Z.; Mousavi, S.V.; Talachi, N.; Tafakhori, A.; Aghamollaii, V. Safety and Efficacy of Melatonin, Clonazepam, and Trazodone in Patients with Parkinson’s Disease and Sleep Disorders: A Randomized, Double-Blind Trial. Neurol. Sci. 2022, 43, 6141–6148. [Google Scholar] [CrossRef]
- Delgado-Lara, D.L.; González-Enríquez, G.V.; Torres-Mendoza, B.M.; González-Usigli, H.; Cárdenas-Bedoya, J.; Macías-Islas, M.A.; de la Rosa, A.C.; Jiménez-Delgado, A.; Pacheco-Moisés, F.; Cruz-Serrano, J.A.; et al. Effect of Melatonin Administration on the PER1 and BMAL1 Clock Genes in Patients with Parkinson’s Disease. Biomed. Pharmacother. 2020, 129, 110485. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Delgado, A.; Ortiz, G.G.; Delgado-Lara, D.L.; González-Usigli, H.A.; González-Ortiz, L.J.; Cid-Hernández, M.; Cruz-Serrano, J.A.; Pacheco-Moisés, F.P. Effect of Melatonin Administration on Mitochondrial Activity and Oxidative Stress Markers in Patients with Parkinson’s Disease. Oxid. Med. Cell. Longev. 2021, 2021, 5577541. [Google Scholar] [CrossRef] [PubMed]
- Wongprayoon, P.; Govitrapong, P. Melatonin as a Mitochondrial Protector in Neurodegenerative Diseases. Cell. Mol. Life Sci. 2017, 74, 3999–4014. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, B.S. The Neuroprotective Role of Melatonin in Neurological Disorders. J. Neurosci. Res. 2018, 96, 1136–1149. [Google Scholar] [CrossRef]
- Liu, J.; Clough, S.J.; Hutchinson, A.J.; Adamah-Biassi, E.B.; Popovska-Gorevski, M.; Dubocovich, M.L. MT 1 and MT 2 Melatonin Receptors: A Therapeutic Perspective. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 361–383. [Google Scholar] [CrossRef]
- Videnovic, A.; Willis, G.L. Circadian System—A Novel Diagnostic and Therapeutic Target in Parkinson’s Disease? Mov. Disord. 2016, 31, 260–269. [Google Scholar] [CrossRef]
- Tamtaji, O.R.; Reiter, R.J.; Alipoor, R.; Dadgostar, E.; Kouchaki, E.; Asemi, Z. Melatonin and Parkinson Disease: Current Status and Future Perspectives for Molecular Mechanisms. Cell. Mol. Neurobiol. 2020, 40, 15–23. [Google Scholar] [CrossRef]
- Ma, H.; Yan, J.; Sun, W.; Jiang, M.; Zhang, Y. Melatonin Treatment for Sleep Disorders in Parkinson’s Disease: A Meta-Analysis and Systematic Review. Front. Aging Neurosci. 2022, 14, 784314. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Wang, X.; Liu, H.; Wang, T.; Lin, Z.; Xiong, N. Neuroprotective Effect of Melatonin on Sleep Disorders Associated with Parkinson’s Disease. Antioxidants 2023, 12, 396. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional Cloning of the Mouse Obese Gene and Its Human Homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Palhinha, L.; Liechocki, S.; Hottz, E.D.; da Pereira, J.A.S.; de Almeida, C.J.; Moraes-Vieira, P.M.M.; Bozza, P.T.; Maya-Monteiro, C.M. Leptin Induces Proadipogenic and Proinflammatory Signaling in Adipocytes. Front. Endocrinol. 2019, 10, 841. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Zhong, L.; Zhu, C.; Zhao, H.; Zhao, F.; Cui, R.; Gao, S.; Li, B. Role of Leptin in Mood Disorder and Neurodegenerative Disease. Front. Neurosci. 2019, 13, 378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chua, S., Jr. Leptin Function and Regulation. Compr. Physiol. 2017, 8, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Signore, A.P.; Zhang, F.; Weng, Z.; Gao, Y.; Chen, J. Leptin Neuroprotection in the CNS: Mechanisms and Therapeutic Potentials. J. Neurochem. 2008, 106, 1977–1990. [Google Scholar] [CrossRef] [PubMed]
- Lorefält, B.; Toss, G.; Granérus, A.-K. Weight Loss, Body Fat Mass, and Leptin in Parkinson’s Disease. Mov. Disord. 2009, 24, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Regensburger, M.; Rasul Chaudhry, S.; Yasin, H.; Zhao, Y.; Stadlbauer, A.; Buchfelder, M.; Kinfe, T. Emerging Roles of Leptin in Parkinson’s Disease: Chronic Inflammation, Neuroprotection and More? Brain. Behav. Immun. 2023, 107, 53–61. [Google Scholar] [CrossRef]
- Rocha, N.P.; Scalzo, P.L.; Barbosa, I.G.; de Sousa, M.S.; Morato, I.B.; Vieira, É.L.M.; Christo, P.P.; Reis, H.J.; Teixeira, A.L. Circulating Levels of Adipokines in Parkinson’s Disease. J. Neurol. Sci. 2014, 339, 64–68. [Google Scholar] [CrossRef]
- Acquarone, E.; Monacelli, F.; Borghi, R.; Nencioni, A.; Odetti, P. Resistin: A Reappraisal. Mech. Ageing Dev. 2019, 178, 46–63. [Google Scholar] [CrossRef]
- Lu, D.-Y.; Chen, J.-H.; Tan, T.-W.; Huang, C.-Y.; Yeh, W.-L.; Hsu, H.-C. Resistin Protects against 6-Hydroxydopamine-Induced Cell Death in Dopaminergic-like MES23.5 Cells. J. Cell. Physiol. 2013, 228, 563–571. [Google Scholar] [CrossRef]
- Filková, M.; Haluzík, M.; Gay, S.; Šenolt, L. The Role of Resistin as a Regulator of Inflammation: Implications for Various Human Pathologies. Clin. Immunol. 2009, 133, 157–170. [Google Scholar] [CrossRef]
- Codoñer-Franch, P.; Alonso-Iglesias, E. Resistin: Insulin Resistance to Malignancy. Clin. Chim. Acta 2015, 438, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Jamaluddin, M.S.; Yan, S.; Lü, J.; Liang, Z.; Yao, Q.; Chen, C. Resistin Increases Monolayer Permeability of Human Coronary Artery Endothelial Cells. PLoS ONE 2013, 8, e84576. [Google Scholar] [CrossRef] [PubMed]
- Sardi, F.; Fassina, L.; Venturini, L.; Inguscio, M.; Guerriero, F.; Rolfo, E.; Ricevuti, G. Alzheimer’s Disease, Autoimmunity and Inflammation. The Good, the Bad and the Ugly. Autoimmun. Rev. 2011, 11, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Silswal, N.; Singh, A.K.; Aruna, B.; Mukhopadhyay, S.; Ghosh, S.; Ehtesham, N.Z. Human Resistin Stimulates the Pro-Inflammatory Cytokines TNF-α and IL-12 in Macrophages by NF-ΚB-Dependent Pathway. Biochem. Biophys. Res. Commun. 2005, 334, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Xiaoying, L.; Li, T.; Yu, S.; Jiusheng, J.; Jilin, Z.; Jiayi, W.; Dongxin, L.; Wengang, F.; Xinyue, Z.; Hao, Y.; et al. Resistin-Inhibited Neural Stem Cell-Derived Astrocyte Differentiation Contributes to Permeability Destruction of the Blood–Brain Barrier. Neurochem. Res. 2019, 44, 905–916. [Google Scholar] [CrossRef]
- Bruun, J.M.; Lihn, A.S.; Verdich, C.; Pedersen, S.B.; Toubro, S.; Astrup, A.; Richelsen, B. Regulation of Adiponectin by Adipose Tissue-Derived Cytokines: In Vivo and In Vitro Investigations in Humans. Am. J. Physiol. Metab. 2003, 285, E527–E533. [Google Scholar] [CrossRef]
- Cnop, M.; Havel, P.J.; Utzschneider, K.M.; Carr, D.B.; Sinha, M.K.; Boyko, E.J.; Retzlaff, B.M.; Knopp, R.H.; Brunzell, J.D.; Kahn, S.E. Relationship of Adiponectin to Body Fat Distribution, Insulin Sensitivity and Plasma Lipoproteins: Evidence for Independent Roles of Age and Sex. Diabetologia 2003, 46, 459–469. [Google Scholar] [CrossRef]
- Kadowaki, T. Adiponectin and Adiponectin Receptors in Insulin Resistance, Diabetes, and the Metabolic Syndrome. J. Clin. Investig. 2006, 116, 1784–1792. [Google Scholar] [CrossRef]
- Gavrila, A.; Chan, J.L.; Yiannakouris, N.; Kontogianni, M.; Miller, L.C.; Orlova, C.; Mantzoros, C.S. Serum Adiponectin Levels Are Inversely Associated with Overall and Central Fat Distribution but Are Not Directly Regulated by Acute Fasting or Leptin Administration in Humans: Cross-Sectional and Interventional Studies. J. Clin. Endocrinol. Metab. 2003, 88, 4823–4831. [Google Scholar] [CrossRef]
- Halleux, C.M.; Takahashi, M.; Delporte, M.L.; Detry, R.; Funahashi, T.; Matsuzawa, Y.; Brichard, S.M. Secretion of Adiponectin and Regulation of ApM1 Gene Expression in Human Visceral Adipose Tissue. Biochem. Biophys. Res. Commun. 2001, 288, 1102–1107. [Google Scholar] [CrossRef]
- Keller, P.; Møller, K.; Krabbe, K.S.; Pedersen, B.K. Circulating Adiponectin Levels during Human Endotoxaemia. Clin. Exp. Immunol. 2003, 134, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Lihn, A.S.; Pedersen, S.B.; Richelsen, B. Adiponectin: Action, Regulation and Association to Insulin Sensitivity. Obes. Rev. 2005, 6, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Polito, R.; Di Meo, I.; Barbieri, M.; Daniele, A.; Paolisso, G.; Rizzo, M.R. Adiponectin Role in Neurodegenerative Diseases: Focus on Nutrition Review. Int. J. Mol. Sci. 2020, 21, 9255. [Google Scholar] [CrossRef] [PubMed]
- Haugen, F.; Drevon, C.A. Activation of Nuclear Factor-ΚB by High Molecular Weight and Globular Adiponectin. Endocrinology 2007, 148, 5478–5486. [Google Scholar] [CrossRef]
- Beitz, J.M. Parkinson s Disease a Review. Front. Biosci. 2014, S6, S415. [Google Scholar] [CrossRef]
- Bloemer, J.; Pinky, P.D.; Govindarajulu, M.; Hong, H.; Judd, R.; Amin, R.H.; Moore, T.; Dhanasekaran, M.; Reed, M.N.; Suppiramaniam, V. Role of Adiponectin in Central Nervous System Disorders. Neural Plast. 2018, 2018, 4593530. [Google Scholar] [CrossRef]
- Santaella, A.; Kuiperij, H.B.; van Rumund, A.; Esselink, R.A.J.; van Gool, A.J.; Bloem, B.R.; Verbeek, M.M. Inflammation Biomarker Discovery in Parkinson’s Disease and Atypical Parkinsonisms. BMC Neurol. 2020, 20, 26. [Google Scholar] [CrossRef]
- Kim, J.-Y.; van de Wall, E.; Laplante, M.; Azzara, A.; Trujillo, M.E.; Hofmann, S.M.; Schraw, T.; Durand, J.L.; Li, H.; Li, G.; et al. Obesity-Associated Improvements in Metabolic Profile through Expansion of Adipose Tissue. J. Clin. Investig. 2007, 117, 2621–2637. [Google Scholar] [CrossRef]
- Chinta, S.J.; Lieu, C.A.; DeMaria, M.; Laberge, R.-M.; Campisi, J.; Andersen, J.K. Environmental Stress, Ageing and Glial Cell Senescence: A Novel Mechanistic Link to Parkinson’s Disease? J. Intern. Med. 2013, 273, 429–436. [Google Scholar] [CrossRef]
- Hoehn, M.M.; Yahr, M.D. Parkinsonism: Onset, Progression, and Mortality. Neurology 1967, 17, 427. [Google Scholar] [CrossRef]
- Mańka, S.; Baj, Z.; Majewska, E. The Influence of Melatonin on Apoptosis of Human Neutrophils. Postep. Hig. Med. Dosw. 2019, 73, 81–91. [Google Scholar] [CrossRef]
- Cipolla-Neto, J.; Amaral, F.G.D. Melatonin as a Hormone: New Physiological and Clinical Insights. Endocr. Rev. 2018, 39, 990–1028. [Google Scholar] [CrossRef] [PubMed]
- Vural, E.M.S.; van Munster, B.C.; de Rooij, S.E. Optimal Dosages for Melatonin Supplementation Therapy in Older Adults: A Systematic Review of Current Literature. Drugs Aging 2014, 31, 441–451. [Google Scholar] [CrossRef] [PubMed]
- del Valle Bessone, C.; Fajreldines, H.D.; de Barboza, G.E.D.; Tolosa de Talamoni, N.G.; Allemandi, D.A.; Carpentieri, A.R.; Quinteros, D.A. Protective Role of Melatonin on Retinal Ganglionar Cell: In Vitro an in Vivo Evidences. Life Sci. 2019, 218, 233–240. [Google Scholar] [CrossRef]
- Cardinali, D.P. Melatonin: Clinical Perspectives in Neurodegeneration. Front. Endocrinol. 2019, 10, 480. [Google Scholar] [CrossRef] [PubMed]
- Grivas, T.B.; Savvidou, O.D. Melatonin the “Light of Night” in Human Biology and Adolescent Idiopathic Scoliosis. Scoliosis 2007, 2, 6. [Google Scholar] [CrossRef]
- Scholtens, R.M.; van Munster, B.C.; van Kempen, M.F.; de Rooij, S.E.J.A. Physiological Melatonin Levels in Healthy Older People: A Systematic Review. J. Psychosom. Res. 2016, 86, 20–27. [Google Scholar] [CrossRef]
- Xie, S.; Fan, W.; He, H.; Huang, F. Role of Melatonin in the Regulation of Pain. J. Pain Res. 2020, 13, 331–343. [Google Scholar] [CrossRef]
- Tchekalarova, J.; Tzoneva, R. Oxidative Stress and Aging as Risk Factors for Alzheimer’s Disease and Parkinson’s Disease: The Role of the Antioxidant Melatonin. Int. J. Mol. Sci. 2023, 24, 3022. [Google Scholar] [CrossRef]
- Breen, D.P.; Nombela, C.; Vuono, R.; Jones, P.S.; Fisher, K.; Burn, D.J.; Brooks, D.J.; Reddy, A.B.; Rowe, J.B.; Barker, R.A. Hypothalamic Volume Loss Is Associated with Reduced Melatonin Output in Parkinson’s Disease. Mov. Disord. 2016, 31, 1062–1066. [Google Scholar] [CrossRef]
- Gunata, M.; Parlakpinar, H.; Acet, H.A. Melatonin: A Review of Its Potential Functions and Effects on Neurological Diseases. Rev. Neurol. 2020, 176, 148–165. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Du, Y.; Yuan, S.; Shen, J.; Lin, X.; Zheng, Z. Serum Melatonin Is an Alternative Index of Parkinson’s Disease Severity. Brain Res. 2014, 1547, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Videnovic, A.; Golombek, D. Circadian Dysregulation in Parkinson’s Disease. Neurobiol. Sleep Circadian Rhythm. 2017, 2, 53–58. [Google Scholar] [CrossRef]
- Kataoka, H.; Saeki, K.; Kurumatani, N.; Sugie, K.; Obayashi, K. Melatonin Secretion in Patients with Parkinson’s Disease Receiving Different-Dose Levodopa Therapy. Sleep Med. 2020, 75, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Bolitho, S.J.; Naismith, S.L.; Rajaratnam, S.M.W.; Grunstein, R.R.; Hodges, J.R.; Terpening, Z.; Rogers, N.; Lewis, S.J.G. Disturbances in Melatonin Secretion and Circadian Sleep–Wake Regulation in Parkinson Disease. Sleep Med. 2014, 15, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Durakoglugil, M.; Irving, A.J.; Harvey, J. Leptin Induces a Novel Form of NMDA Receptor-Dependent Long-Term Depression. J. Neurochem. 2005, 95, 396–405. [Google Scholar] [CrossRef]
- Ozdilek, B.; Kenangil, G. Serum Leptin Concentrations in Turkish Parkinson’s Disease Population. Parkinsons. Dis. 2014, 2014, 576020. [Google Scholar] [CrossRef]
- Fiszer, U.; Michałowska, M.; Baranowska, B.; Wolińska-Witort, E.; Jeske, W.; Jethon, M.; Piaścik-Gromada, M.; Marcinowska-Suchowierska, E. Leptin and Ghrelin Concentrations and Weight Loss in Parkinson’s Disease. Acta Neurol. Scand. 2010, 121, 230–236. [Google Scholar] [CrossRef]
- Evidente, V.G.H.; Caviness, J.N.; Adler, C.H.; Gwinn-Hardy, K.A.; Pratley, R.E. Serum Leptin Concentrations and Satiety in Parkinson’s Disease Patients with and without Weight Loss. Mov. Disord. 2001, 16, 924–927. [Google Scholar] [CrossRef]
- Markaki, E.; Ellul, J.; Kefalopoulou, Z.; Trachani, E.; Theodoropoulou, A.; Kyriazopoulou, V.; Constantoyannis, C. The Role of Ghrelin, Neuropeptide Y and Leptin Peptides in Weight Gain after Deep Brain Stimulation for Parkinson’s Disease. Stereotact. Funct. Neurosurg. 2012, 90, 104–112. [Google Scholar] [CrossRef]
- Adams, F.; Boschmann, M.; Lobsien, E.; Kupsch, A.; Lipp, A.; Franke, G.; Leisse, M.C.; Janke, J.; Gottschalk, S.; Spranger, J.; et al. Influences of Levodopa on Adipose Tissue and Skeletal Muscle Metabolism in Patients with Idiopathic Parkinson’s Disease. Eur. J. Clin. Pharmacol. 2008, 64, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, C.G.; Trenkwalder, C. Body Weight in Patients with Parkinson’s Disease. Mov. Disord. 2006, 21, 1824–1830. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Park, K.-H.; Cho, Y.M.; Chung, S.S.; Cho, H.J.; Cho, S.Y.; Kim, S.J.; Kim, S.Y.; Lee, H.K.; Park, K.S. Resistin Is Secreted from Macrophages in Atheromas and Promotes Atherosclerosis. Cardiovasc. Res. 2006, 69, 76–85. [Google Scholar] [CrossRef] [PubMed]
- McTernan, P.G.; Kusminski, C.M.; Kumar, S. Resistin. Curr. Opin. Lipidol. 2006, 17, 170–175. [Google Scholar] [CrossRef]
- Kusminski, C.M.; Mcternan, P.G.; Kumar, S. Role of Resistin in Obesity, Insulin Resistance and Type II Diabetes. Clin. Sci. 2005, 109, 243–256. [Google Scholar] [CrossRef]
- De Rui, M.; Inelmen, E.M.; Trevisan, C.; Pigozzo, S.; Manzato, E.; Sergi, G. Parkinson’s Disease and the Non-Motor Symptoms: Hyposmia, Weight Loss, Osteosarcopenia. Aging Clin. Exp. Res. 2020, 32, 1211–1218. [Google Scholar] [CrossRef]
- Jenner, P. Molecular Mechanisms of L-DOPA-Induced Dyskinesia. Nat. Rev. Neurosci. 2008, 9, 665–677. [Google Scholar] [CrossRef]
- Kataoka, H.; Sugie, K. Serum Adiponectin Levels between Patients with Parkinson’s Disease and Those with PSP. Neurol. Sci. 2020, 41, 1125–1131. [Google Scholar] [CrossRef]
- Sekiyama, K.; Waragai, M.; Akatsu, H.; Sugama, S.; Takenouchi, T.; Takamatsu, Y.; Fujita, M.; Sekigawa, A.; Rockenstein, E.; Inoue, S.; et al. Disease Modifying Effect of Adiponectin in Model of α-synucleinopathies. Ann. Clin. Transl. Neurol. 2014, 1, 479–489. [Google Scholar] [CrossRef]
- Sharma, J.C.; Bachmann, C.G.; Linazasoro, G. Classifying Risk Factors for Dyskinesia in Parkinson’s Disease. Park. Relat. Disord. 2010, 16, 490–497. [Google Scholar] [CrossRef]
- Sharma, J.C.; Macnamara, L.; Hasoon, M.; Vassallo, M.; Ross, I. Cascade of Levodopa Dose and Weight-Related Dyskinesia in Parkinson’s Disease (LD–WD-PD Cascade). Park. Relat. Disord. 2006, 12, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Cassani, E.; Cancello, R.; Cavanna, F.; Maestrini, S.; Di Blasio, A.M.; Liuzzi, A.; Pezzoli, G.; Barichella, M. Serum Adiponectin Levels in Advanced-Stage Parkinson’s Disease Patients. Park. Dis. 2011, 2011, 624764. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
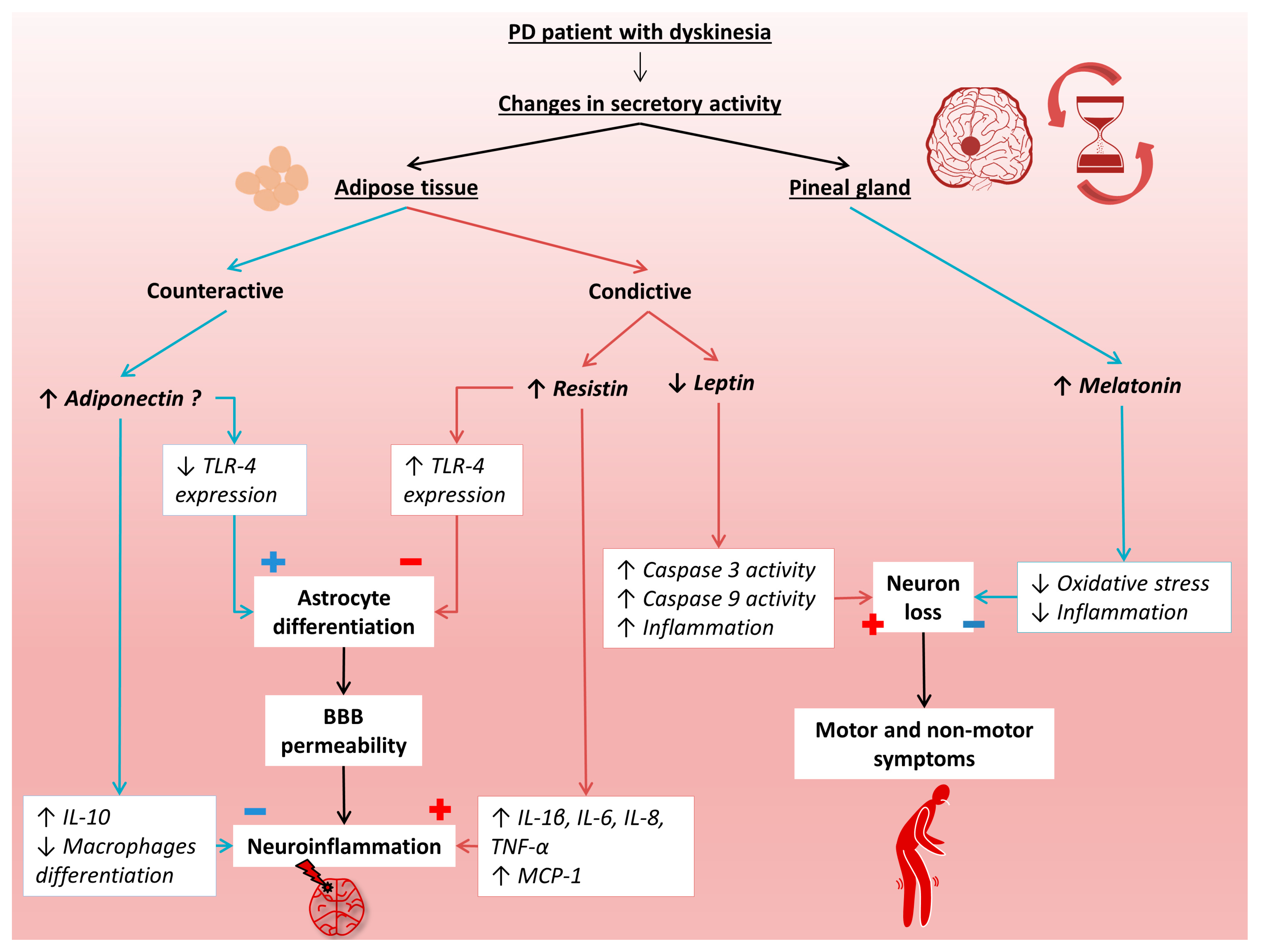{kind=link}
| Parameter | Parkinson’s Disease | Control | p Value | Power of a Test | ||
|---|---|---|---|---|---|---|
| No Dyskinesia | With Dyskinesia | |||||
| n (Female/Male) | 20 (9/11) | 24 (10/14) | 20 (8/12) | - | - | |
| Age [years] | Mean | 68.250 | 68.417 | 66.950 | 0.6220 | 0.1069 |
| SEM | 1.326 | 1.302 | 0.540 | |||
| Median | 69.000 | 68.500 | 67.500 | |||
| IQR | 7.250 | 6.500 | 4.000 | |||
| Body mass [kg] | Mean | 81.250 | 74.021 | 78.500 | 0.7065 | 1.0000 |
| SEM | 4.852 | 3.147 | 2.162 | |||
| Median | 78.500 | 74.000 | 77.500 | |||
| IQR | 21.500 | 14.500 | 15.250 | |||
| Height [cm] | Mean | 170.011 | 166.958 | 169.700 | 0.2443 | 0.2374 |
| SEM | 1.761 | 1.372 | 1.093 | |||
| Median | 169.000 | 167.500 | 169.000 | |||
| IQR | 10.000 | 6.500 | 6.250 | |||
| BMI [kg/m2] | Mean | 27.829 | 26.419 | 27.218 | 0.4982 | 0.9604 |
| SEM | 1.214 | 1.006 | 0.608 | |||
| Median | 26.037 | 25.510 | 27.184 | |||
| IQR | 4.183 | 4.412 | 3.899 | |||
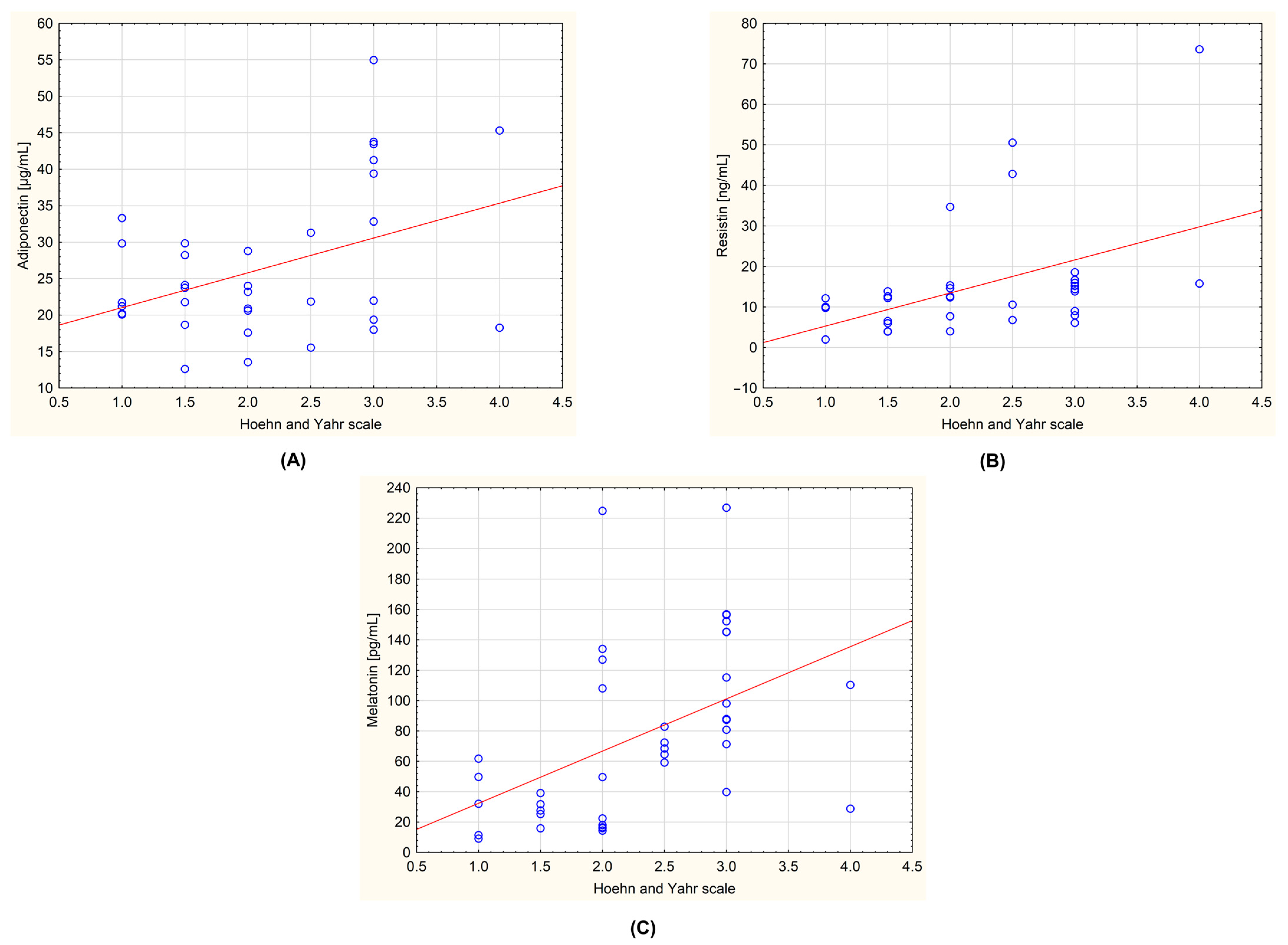
| Hoehn–Yahr scale | Mean | 1.500 | 2.813 | 0.000 | <0.0001 | 0.9999 |
| SEM | 0.089 | 0.108 | 0.000 | |||
| Median | 1.500 | 3.000 | 0.000 | |||
| IQR | 1.000 | 0.500 | 0.000 | |||
| Years since diagnosis [years] | Mean | 1.300 | 8.167 | 0.000 | <0.0001 | 1.0000 |
| SEM | 0.105 | 0.354 | 0.000 | |||
| Median | 1.000 | 8.000 | 0.000 | |||
| IQR | 1.000 | 3.250 | 0.000 | |||
| Drugs, Dose | PD No Dyskinesia [n = 20] | PD with Dyskinesia [n = 24] |
|---|---|---|
| no treatment | 0 | 0 |
| madopar HBS | 14 | 12 |
| madopar 250 + HBS | 0 | 7 |
| madopar HBS + madopar 125 | 0 | 3 |
| madopar HBS + madopar 125 + madopar 625 | 1 | 1 |
| madopar 625 + HBS | 4 | 0 |
| madopar 125 + madopar 125 | 1 | 0 |
| madopar x4 | 0 | 1 |
| Parameter | Parkinson’s Disease | Control | p Value | Power of a Test | ||
|---|---|---|---|---|---|---|
| No Dyskinesia | With Dyskinesia | |||||
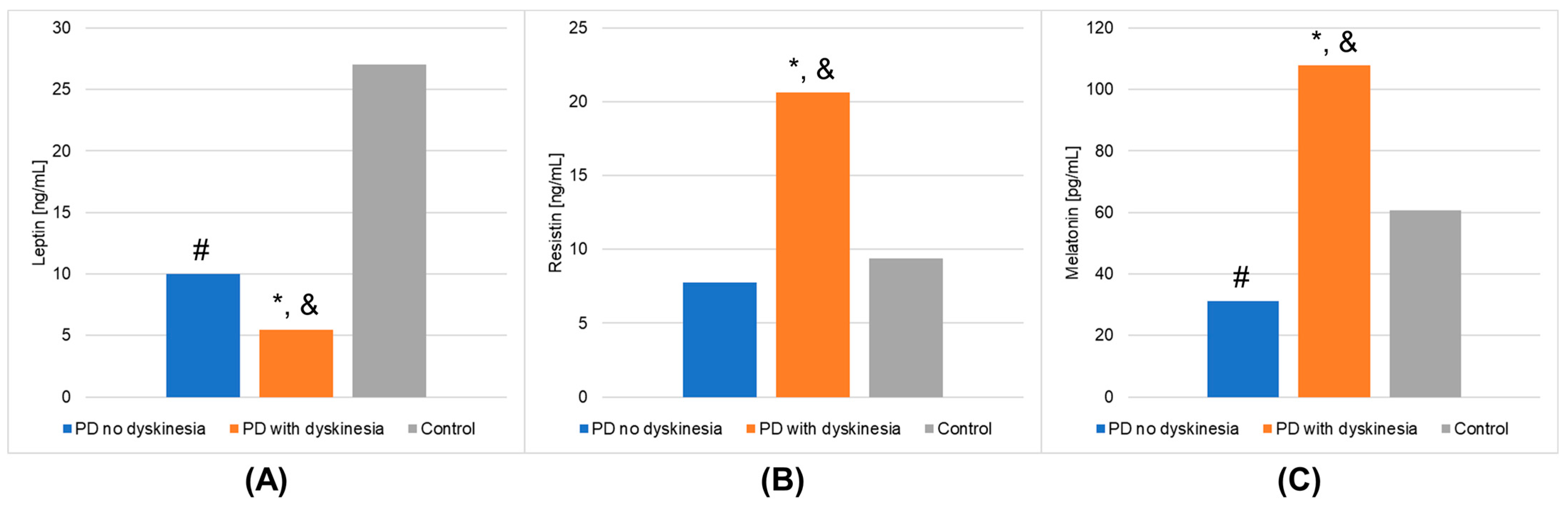
| Adiponectin [µg/mL] | Mean | 22.833 | 29.778 | 29.082 | 0.0626 | 0.9998 |
| SEM | 1.486 | 2.824 | 1.981 | |||
| Median | 21.753 | 23.595 | 30.513 | |||
| IQR | 8.638 | 20.103 | 7.331 | |||
| Leptin [ng/mL] | Mean | 9.983 | 5.431 | 27.030 | <0.0001 | 1.0000 |
| SEM | 1.414 | 0.933 | 2.860 | |||
| Median | 8.725 | 4.070 | 26.664 | |||
| IQR | 7.995 | 4.076 | 19.910 | |||
| Resistin [ng/mL] | Mean | 7.768 | 20.645 | 9.392 | 0.0013 | 0.9082 |
| SEM | 1.145 | 4.025 | 0.993 | |||
| Median | 7.140 | 14.640 | 9.143 | |||
| IQR | 8.188 | 6.159 | 4.724 | |||
| Melatonin [pg/mL] | Mean | 31.205 | 107.742 | 60.580 | <0.0001 | 0.9997 |
| SEM | 6.256 | 10.719 | 4.844 | |||
| Median | 23.841 | 93.025 | 60.292 | |||
| IQR | 17.858 | 74.497 | 31.985 | |||
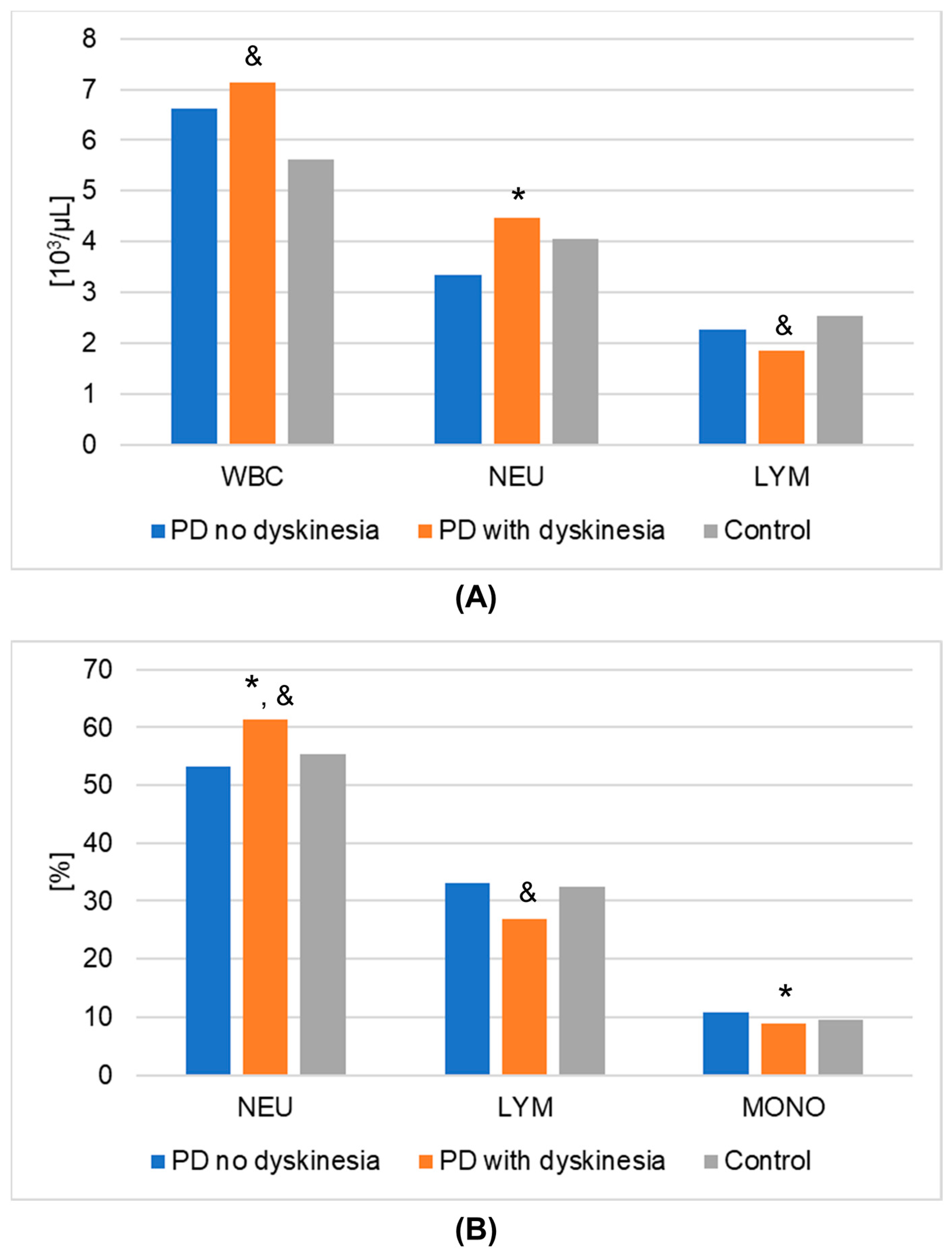
| WBC [103/µL] | Mean | 6.635 | 7.150 | 5.625 | 0.0245 | 0.5693 |
| SEM | 0.511 | 0.388 | 0.211 | |||
| Median | 6.570 | 6.725 | 5.350 | |||
| IQR | 2.195 | 2.025 | 1.475 | |||
| RBC [106/μL] | Mean | 4.641 | 4.622 | 4.382 | 0.1117 | 0.3545 |
| SEM | 0.097 | 0.093 | 0.090 | |||
| Median | 4.715 | 4.620 | 4.370 | |||
| IQR | 0.480 | 0.762 | 0.283 | |||
| Hb [g/dL] | Mean | 14.225 | 14.154 | 13.865 | 0.6297 | 0.1053 |
| SEM | 0.232 | 0.295 | 0.273 | |||
| Median | 14.500 | 14.000 | 14.100 | |||
| IQR | 1.425 | 2.025 | 1.600 | |||
| HCT [%] | Mean | 42.090 | 40.421 | 40.560 | 0.1383 | 0.9929 |
| SEM | 0.686 | 1.747 | 0.396 | |||
| Median | 42.850 | 41.150 | 40.200 | |||
| IQR | 4.475 | 4.800 | 1.375 | |||
| MCV [fL] | Mean | 90.915 | 89.504 | 90.135 | 0.9075 | 0.9579 |
| SEM | 0.817 | 2.013 | 1.000 | |||
| Median | 91.000 | 91.900 | 91.300 | |||
| IQR | 3.750 | 5.125 | 4.400 | |||
| MCH [pg] | Mean | 30.700 | 30.588 | 31.050 | 0.3596 | 0.2495 |
| SEM | 0.273 | 0.291 | 0.341 | |||
| Median | 30.700 | 30.700 | 31.200 | |||
| IQR | 1.150 | 1.675 | 1.825 | |||
| MCHC [g/dL] | Mean | 33.765 | 33.471 | 33.767 | 0.1925 | 0.1517 |
| SEM | 0.162 | 0.174 | 0.316 | |||
| Median | 33.700 | 33.400 | 34.055 | |||
| IQR | 0.850 | 0.625 | 1.666 | |||
| RDW [%] | Mean | 13.710 | 14.079 | 14.115 | 0.2986 | 0.2080 |
| SEM | 0.172 | 0.184 | 0.235 | |||
| Median | 13.600 | 13.900 | 14.150 | |||
| IQR | 0.875 | 1.150 | 0.850 | |||
| PLT [103/μL] | Mean | 234.450 | 217.667 | 254.250 | 0.0654 | 0.4336 |
| SEM | 13.984 | 9.871 | 8.587 | |||
| Median | 216.500 | 211.000 | 246.000 | |||
| IQR | 83.500 | 81.750 | 59.750 | |||
| MPV [fL] | Mean | 10.705 | 10.925 | 9.315 | <0.0001 | 0.9889 |
| SEM | 0.201 | 0.221 | 0.212 | |||
| Median | 10.900 | 10.850 | 9.050 | |||
| IQR | 1.250 | 1.375 | 1.150 | |||
| % NEU [%] | Mean | 53.320 | 61.458 | 55.293 | 0.0059 | 0.7305 |
| SEM | 2.446 | 1.669 | 1.242 | |||
| Median | 55.700 | 62.350 | 55.410 | |||
| IQR | 18.425 | 10.800 | 9.389 | |||
| % LYM [%] | Mean | 33.075 | 26.925 | 32.541 | 0.0168 | 0.6166 |
| SEM | 2.157 | 1.555 | 1.186 | |||
| Median | 33.350 | 24.750 | 31.589 | |||
| IQR | 18.800 | 11.450 | 6.511 | |||
| % MONO [%] | Mean | 10.730 | 8.888 | 9.468 | 0.0447 | 0.9950 |
| SEM | 0.647 | 0.615 | 0.431 | |||
| Median | 9.900 | 8.200 | 9.352 | |||
| IQR | 2.850 | 3.225 | 2.967 | |||
| % EOS [%] | Mean | 2.740 | 2.196 | 2.362 | 0.6014 | 0.3214 |
| SEM | 0.355 | 0.260 | 0.275 | |||
| Median | 2.400 | 2.050 | 2.276 | |||
| IQR | 2.450 | 1.100 | 1.444 | |||
| % BASO [%] | Mean | 0.495 | 0.425 | 0.489 | 0.5830 | 0.0548 |
| SEM | 0.080 | 0.069 | 0.057 | |||
| Median | 0.300 | 0.350 | 0.541 | |||
| IQR | 0.450 | 0.325 | 0.426 | |||
| NEU [103/μL] | Mean | 3.348 | 4.477 | 4.058 | 0.0173 | 0.8609 |
| SEM | 0.257 | 0.271 | 0.288 | |||
| Median | 3.200 | 4.485 | 4.271 | |||
| IQR | 1.945 | 1.470 | 2.481 | |||
| LYM [103/μL] | Mean | 2.261 | 1.846 | 2.531 | 0.0064 | 0.7222 |
| SEM | 0.166 | 0.137 | 0.148 | |||
| Median | 2.105 | 1.770 | 2.668 | |||
| IQR | 1.253 | 0.865 | 0.981 | |||
| MONO [103/μL] | Mean | 0.698 | 0.691 | 0.715 | 0.2321 | 0.0505 |
| SEM | 0.043 | 0.080 | 0.051 | |||
| Median | 0.685 | 0.565 | 0.701 | |||
| IQR | 0.230 | 0.248 | 0.315 | |||
| EOS [103/μL] | Mean | 0.187 | 0.256 | 0.208 | 0.3068 | 0.0540 |
| SEM | 0.025 | 0.099 | 0.022 | |||
| Median | 0.210 | 0.140 | 0.214 | |||
| IQR | 0.168 | 0.068 | 0.127 | |||
| BASO [103/μL] | Mean | 0.029 | 0.030 | 0.026 | 0.8760 | 0.0500 |
| SEM | 0.003 | 0.003 | 0.001 | |||
| Median | 0.030 | 0.030 | 0.024 | |||
| IQR | 0.015 | 0.020 | 0.012 | |||
| Parameter | p Value | ||
|---|---|---|---|
| No Dyskinesia vs. with Dyskinesia | No Dyskinesia vs. Control | With Dyskinesia vs. Control | |
| Hoehn–Yahr scale | <0.0001 | <0.0001 | <0.0001 |
| Years since diagnosis | <0.0001 | <0.0001 | <0.0001 |
| Leptin | 0.0360 | <0.0001 | <0.0001 |
| Resistin | 0.0030 | 0.8922 | 0.0064 |
| Melatonin | <0.0001 | 0.0025 | 0.0010 |
| WBC | 0.6085 | 0.1895 | 0.0192 |
| MPV | 1.0000 | 0.0002 | <0.0001 |
| % NEU | 0.0280 | 0.8577 | 0.0151 |
| % LYM | 0.0777 | 0.9951 | 0.0192 |
| % MONO | 0.0455 | 0.6250 | 0.6476 |
| NEU | 0.0120 | 0.2033 | 1.0000 |
| LYM | 0.1262 | 0.4398 | 0.0050 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milanowski, J.; Kozerawski, K.; Falęcka, W.; Dudek, D.; Lisewska, B.; Lisewski, P.; Nuszkiewicz, J.; Wesołowski, R.; Wojtasik, J.; Mila-Kierzenkowska, C.; et al. Changes in the Secretion of Melatonin and Selected Adipokines during the Progression of Parkinson’s Disease—Preliminary Studies. Metabolites 2023, 13, 668. https://doi.org/10.3390/metabo13050668
Milanowski J, Kozerawski K, Falęcka W, Dudek D, Lisewska B, Lisewski P, Nuszkiewicz J, Wesołowski R, Wojtasik J, Mila-Kierzenkowska C, et al. Changes in the Secretion of Melatonin and Selected Adipokines during the Progression of Parkinson’s Disease—Preliminary Studies. Metabolites. 2023; 13(5):668. https://doi.org/10.3390/metabo13050668
Chicago/Turabian StyleMilanowski, Jan, Kamil Kozerawski, Weronika Falęcka, Dominik Dudek, Beata Lisewska, Paweł Lisewski, Jarosław Nuszkiewicz, Roland Wesołowski, Jakub Wojtasik, Celestyna Mila-Kierzenkowska, and et al. 2023. "Changes in the Secretion of Melatonin and Selected Adipokines during the Progression of Parkinson’s Disease—Preliminary Studies" Metabolites 13, no. 5: 668. https://doi.org/10.3390/metabo13050668
APA StyleMilanowski, J., Kozerawski, K., Falęcka, W., Dudek, D., Lisewska, B., Lisewski, P., Nuszkiewicz, J., Wesołowski, R., Wojtasik, J., Mila-Kierzenkowska, C., & Szewczyk-Golec, K. (2023). Changes in the Secretion of Melatonin and Selected Adipokines during the Progression of Parkinson’s Disease—Preliminary Studies. Metabolites, 13(5), 668. https://doi.org/10.3390/metabo13050668