
Influence of Cholesterol on the Regulation of Osteoblast Function


Abstract
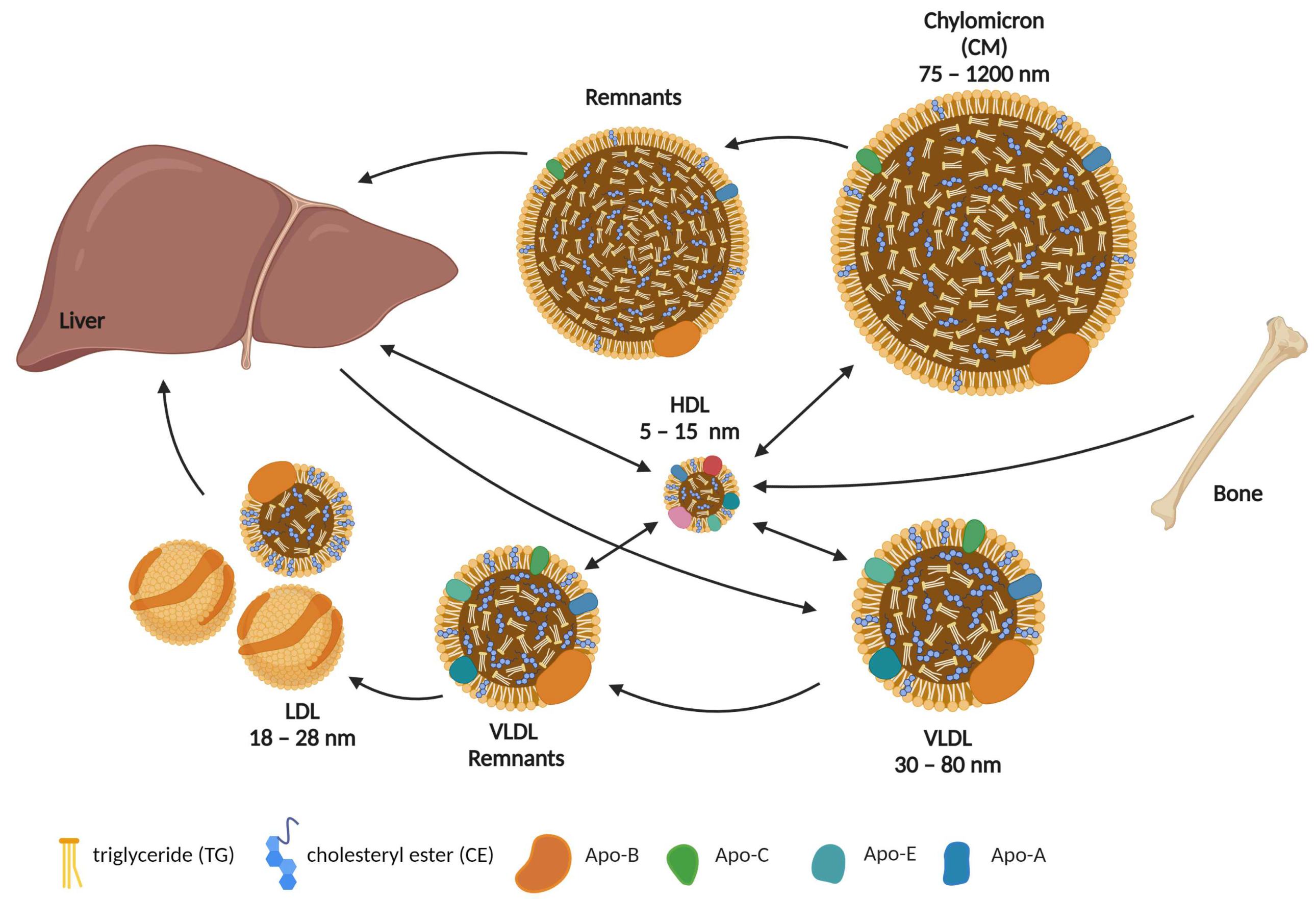
1. Introduction
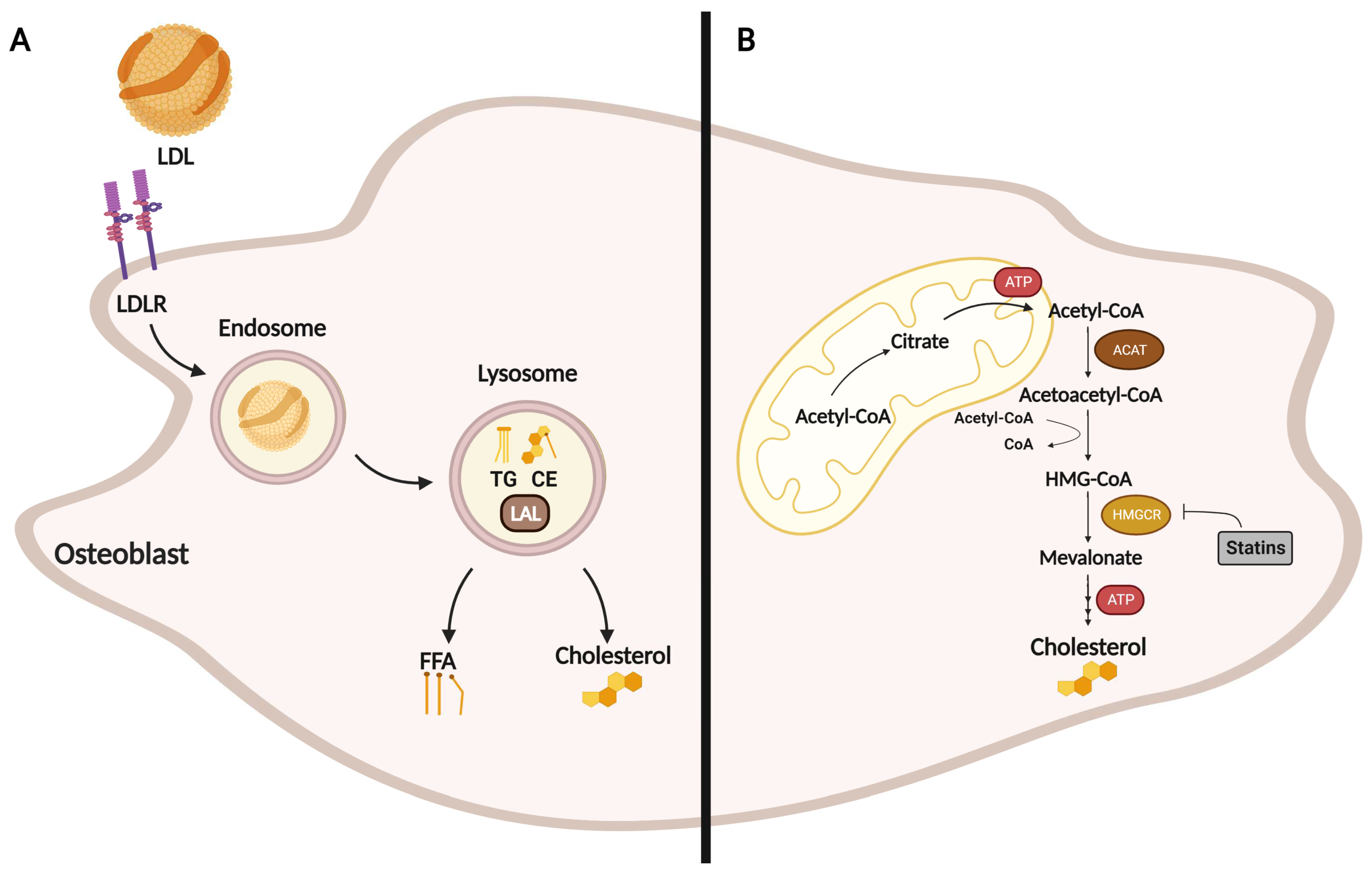
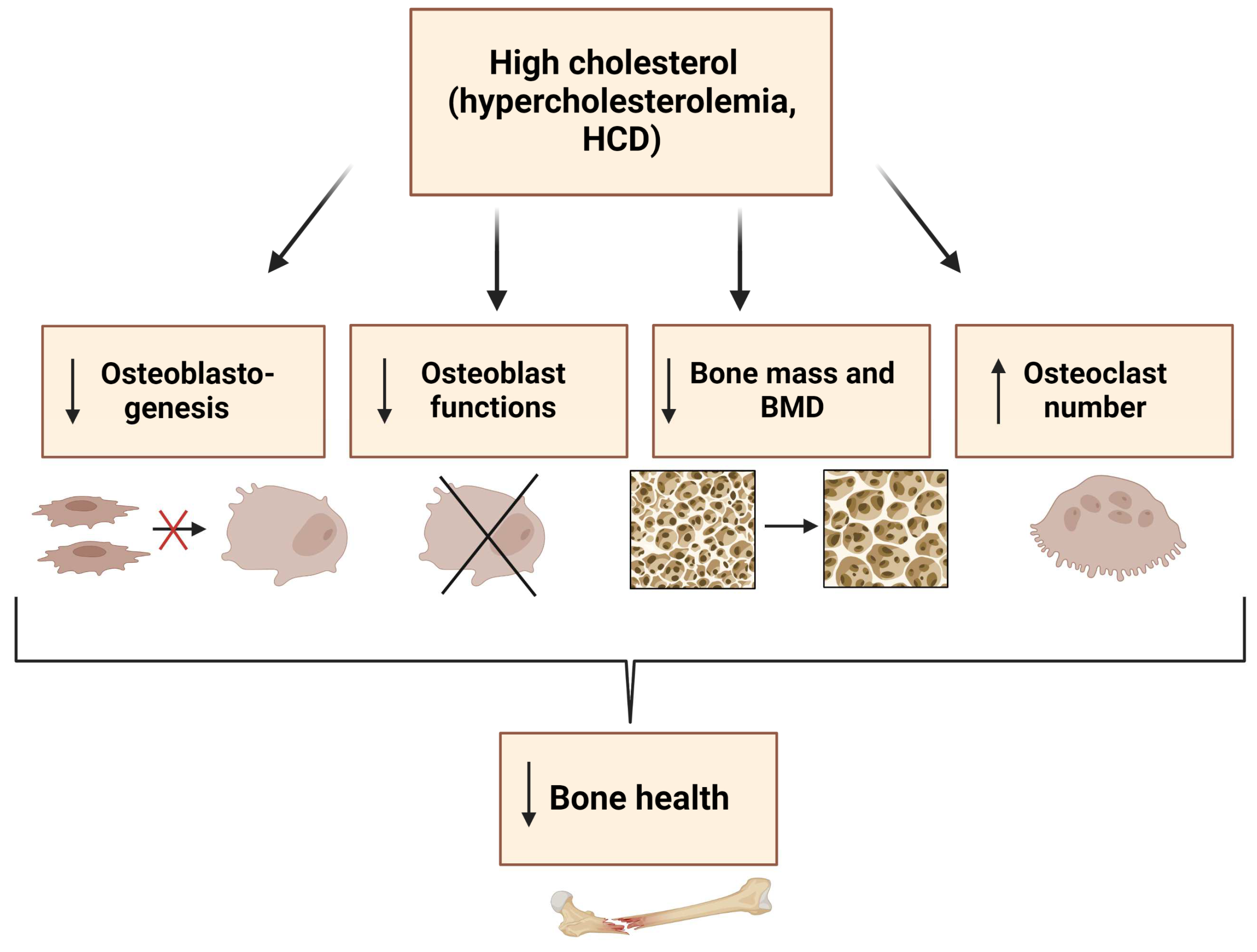
2. Role of Cholesterol in Osteoblast Formation, Function, and Metabolism
3. Effect of Statins on Osteoblasts
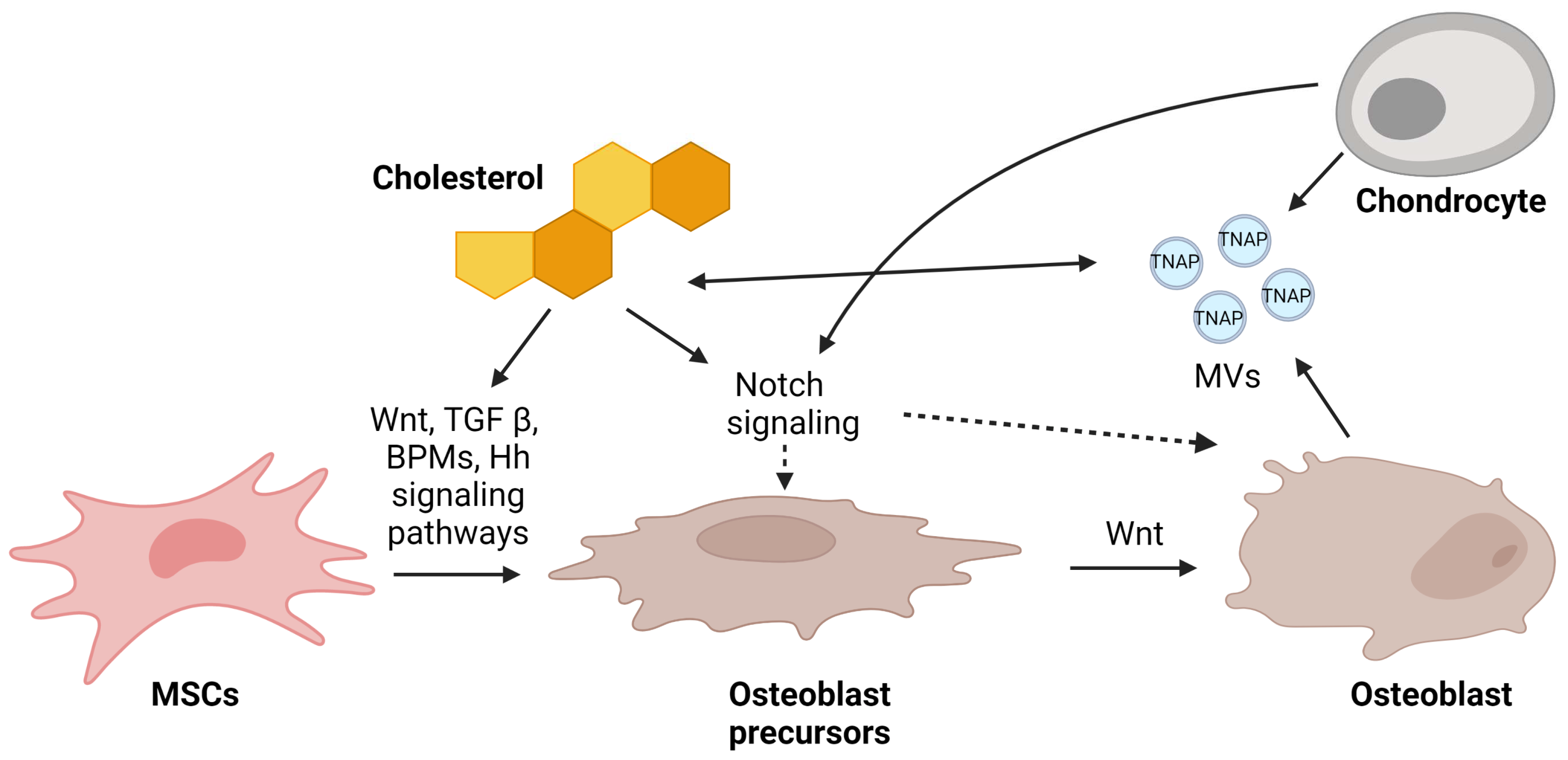
4. Mechanism of Osteoblast Regulation by Cholesterol via Signaling Pathways
4.1. Wnt-Lrp5-β-Catenin
4.2. TGF-β/BMP2
4.3. Notch
4.4. Hedgehog (Hh)
5. Mechanism of Mineralization Regulation by Matrix Vesicles
6. Conclusions
7. Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Su, N.; Yang, J.; Xie, Y.; Du, X.; Chen, H.; Zhou, H.; Chen, L. Bone function, dysfunction and its role in diseases including critical illness. Int. J. Biol. Sci. 2019, 15, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Robling, A.G.; Castillo, A.B.; Turner, C.H. Biomechanical and molecular regulation of bone remodeling. Annu. Rev. Biomed. Eng. 2006, 8, 455–498. [Google Scholar] [CrossRef] [PubMed]
- Florencio-Silva, R.; Sasso, G.R.; Sasso-Cerri, E.; Simoes, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed. Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B. Normal Bone Anatomy and Physiology. Clin. J. Am. Soc. Nephrol. 2008, 3, S131–S139. [Google Scholar] [CrossRef] [PubMed]
- Boskey, A.L. Bone composition: Relationship to bone fragility and antiosteoporotic drug effects. Bonekey Rep. 2013, 2, 447. [Google Scholar] [CrossRef]
- Buckwalter, J.A.; Glimcher, M.J.; Cooper, R.R.; Recker, R. Bone biology. I: Structure, blood supply, cells, matrix, and mineralization. Instr. Course Lect. 1996, 45, 371–386. [Google Scholar]
- Mohan, A.; Girdhar, M.; Kumar, R.; Chaturvedi, H.S.; Vadhel, A.; Solanki, P.R.; Kumar, A.; Kumar, D.; Mamidi, N. Polyhydroxybutyrate-Based Nanocomposites for Bone Tissue Engineering. Pharmaceuticals 2021, 14, 1163. [Google Scholar] [CrossRef]
- Chan, C.K.F.; Gulati, G.S.; Sinha, R.; Tompkins, J.V.; Lopez, M.; Carter, A.C.; Ransom, R.C.; Reinisch, A.; Wearda, T.; Murphy, M.; et al. Identification of the Human Skeletal Stem Cell. Cell 2018, 175, 43–56.e21. [Google Scholar] [CrossRef]
- Marie, P.J. Transcription factors controlling osteoblastogenesis. Arch. Biochem. Biophys. 2008, 473, 98–105. [Google Scholar] [CrossRef]
- Kenkre, J.S.; Bassett, J. The bone remodelling cycle. Ann. Clin. Biochem. 2018, 55, 308–327. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, X.; Bikle, D.D. Osteogenic Differentiation of Periosteal Cells During Fracture Healing. J. Cell. Physiol. 2017, 232, 913–921. [Google Scholar] [CrossRef]
- Franz-Odendaal, T.A.; Hall, B.K.; Witten, P.E. Buried alive: How osteoblasts become osteocytes. Dev. Dyn. 2006, 235, 176–190. [Google Scholar] [CrossRef]
- Brauer, A.; Pohlemann, T.; Metzger, W. Osteogenic differentiation of immature osteoblasts: Interplay of cell culture media and supplements. Biotech. Histochem. 2016, 91, 161–169. [Google Scholar] [CrossRef]
- Dai, R.; Liu, M.; Xiang, X.; Xi, Z.; Xu, H. Osteoblasts and osteoclasts: An important switch of tumour cell dormancy during bone metastasis. J. Exp. Clin. Cancer Res. 2022, 41, 316. [Google Scholar] [CrossRef]
- Blair, H.C.; Larrouture, Q.C.; Li, Y.; Lin, H.; Beer-Stoltz, D.; Liu, L.; Tuan, R.S.; Robinson, L.J.; Schlesinger, P.H.; Nelson, D.J. Osteoblast Differentiation and Bone Matrix Formation In Vivo and In Vitro. Tissue Eng. Part. B. Rev. 2017, 23, 268–280. [Google Scholar] [CrossRef]
- Gordon, J.A.R.; Stein, J.L.; Westendorf, J.J.; van Wijnen, A.J. Chromatin modifiers and histone modifications in bone formation, regeneration, and therapeutic intervention for bone-related disease. Bone 2015, 81, 739–745. [Google Scholar] [CrossRef]
- Yi, S.J.; Lee, H.; Lee, J.; Lee, K.; Kim, J.; Kim, Y.; Park, J.I.; Kim, K. Bone Remodeling: Histone Modifications as Fate Determinants of Bone Cell Differentiation. Int. J. Mol. Sci. 2019, 20, 3147. [Google Scholar] [CrossRef]
- Sen, B.; Xie, Z.; Uzer, G.; Thompson, W.R.; Styner, M.; Wu, X.; Rubin, J. Intranuclear Actin Regulates Osteogenesis. Stem Cells 2015, 33, 3065–3076. [Google Scholar] [CrossRef]
- Suzuki, H.; Tatei, K.; Ohshima, N.; Sato, S.; Izumi, T. Regulation of MC3T3-E1 differentiation by actin cytoskeleton through lipid mediators reflecting the cell differentiation stage. Biochem. Biophys. Res. Commun. 2019, 514, 393–400. [Google Scholar] [CrossRef]
- Ansari, S.; de Wildt, B.W.M.; Vis, M.A.M.; de Korte, C.E.; Ito, K.; Hofmann, S.; Yuana, Y. Matrix Vesicles: Role in Bone Mineralization and Potential Use as Therapeutics. Pharmaceuticals 2021, 14, 289. [Google Scholar] [CrossRef]
- Cui, L.; Houston, D.A.; Farquharson, C.; MacRae, V.E. Characterisation of matrix vesicles in skeletal and soft tissue mineralisation. Bone 2016, 87, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Riddle, R.C.; Clemens, T.L. Bone Cell Bioenergetics and Skeletal Energy Homeostasis. Physiol. Rev. 2017, 97, 667–698. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Xiao, J.; Wang, J.; Ma, Y.; Zhang, Y.; Zhang, Q.; Zhang, Z.; Yin, H. The Interaction Between Intracellular Energy Metabolism and Signaling Pathways During Osteogenesis. Front. Mol. Biosci. 2021, 8, 807487. [Google Scholar] [CrossRef] [PubMed]
- Sautchuk, R., Jr.; Eliseev, R.A. Cell energy metabolism and bone formation. Bone Rep. 2022, 16, 101594. [Google Scholar] [CrossRef] [PubMed]
- Guntur, A.R.; Le, P.T.; Farber, C.R.; Rosen, C.J. Bioenergetics during calvarial osteoblast differentiation reflect strain differences in bone mass. Endocrinology 2014, 155, 1589–1595. [Google Scholar] [CrossRef]
- Ansari, S.; Ito, K.; Hofmann, S. Alkaline Phosphatase Activity of Serum Affects Osteogenic Differentiation Cultures. ACS Omega 2022, 7, 12724–12733. [Google Scholar] [CrossRef]
- Zhang, Q.; Riddle, R.C.; Clemens, T.L. Bone and the regulation of global energy balance. J. Intern. Med. 2015, 277, 681–689. [Google Scholar] [CrossRef]
- Dirckx, N.; Moorer, M.C.; Clemens, T.L.; Riddle, R.C. The role of osteoblasts in energy homeostasis. Nat. Rev. Endocrinol. 2019, 15, 651–665. [Google Scholar] [CrossRef]
- Shum, L.C.; White, N.S.; Mills, B.N.; Bentley, K.L.; Eliseev, R.A. Energy Metabolism in Mesenchymal Stem Cells During Osteogenic Differentiation. Stem Cells Dev. 2016, 25, 114–122. [Google Scholar] [CrossRef]
- Motyl, K.J.; Guntur, A.R.; Carvalho, A.L.; Rosen, C.J. Energy Metabolism of Bone. Toxicol. Pathol. 2017, 45, 887–893. [Google Scholar] [CrossRef]
- Jayapalan, S.; Nandy, A.; Rendina-Ruedy, E. Using Real-Time Cell Metabolic Flux Analyzer to Monitor Osteoblast Bioenergetics. J. Vis. Exp. 2022, e63142. [Google Scholar] [CrossRef]
- Misra, B.B.; Jayapalan, S.; Richards, A.K.; Helderman, R.C.M.; Rendina-Ruedy, E. Untargeted metabolomics in primary murine bone marrow stromal cells reveals distinct profile throughout osteoblast differentiation. Metabolomics 2021, 17, 86. [Google Scholar] [CrossRef]
- Tencerova, M.; Rendina-Ruedy, E.; Neess, D.; Faergeman, N.; Figeac, F.; Ali, D.; Danielsen, M.; Haakonsson, A.; Rosen, C.J.; Kassem, M. Metabolic programming determines the lineage-differentiation fate of murine bone marrow stromal progenitor cells. Bone Res. 2019, 7, 35. [Google Scholar] [CrossRef]
- Guntur, A.R.; Gerencser, A.A.; Le, P.T.; DeMambro, V.E.; Bornstein, S.A.; Mookerjee, S.A.; Maridas, D.E.; Clemmons, D.E.; Brand, M.D.; Rosen, C.J. Osteoblast-like MC3T3-E1 Cells Prefer Glycolysis for ATP Production but Adipocyte-like 3T3-L1 Cells Prefer Oxidative Phosphorylation. J. Bone Miner. Res. 2018, 33, 1052–1065. [Google Scholar] [CrossRef]
- Chen, C.T.; Shih, Y.R.; Kuo, T.K.; Lee, O.K.; Wei, Y.H. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 2008, 26, 960–968. [Google Scholar] [CrossRef]
- Frey, J.L.; Li, Z.; Ellis, J.M.; Zhang, Q.; Farber, C.R.; Aja, S.; Wolfgang, M.J.; Clemens, T.L.; Riddle, R.C. Wnt-Lrp5 signaling regulates fatty acid metabolism in the osteoblast. Mol. Cell. Biol. 2015, 35, 1979–1991. [Google Scholar] [CrossRef]
- Shen, L.; Hu, G.; Karner, C.M. Bioenergetic Metabolism In Osteoblast Differentiation. Curr. Osteoporos. Rep. 2022, 20, 53–64. [Google Scholar] [CrossRef]
- Zampelas, A.; Magriplis, E. New Insights into Cholesterol Functions: A Friend or an Enemy? Nutrients 2019, 11, 1645. [Google Scholar] [CrossRef]
- During, A.; Penel, G.; Hardouin, P. Understanding the local actions of lipids in bone physiology. Prog. Lipid Res. 2015, 59, 126–146. [Google Scholar] [CrossRef]
- Duan, Y.; Gong, K.; Xu, S.; Zhang, F.; Meng, X.; Han, J. Regulation of cholesterol homeostasis in health and diseases: From mechanisms to targeted therapeutics. Signal. Transduct. Target. Ther. 2022, 7, 265. [Google Scholar] [CrossRef]
- Osborne, J.C.; Brewer, H.B. The Plasma Lipoproteins. In Advances in Protein Chemistry; Academic Press: Cambridge, MA, USA, 1977; pp. 253–337. [Google Scholar]
- Parhami, F.; Mody, N.; Gharavi, N.; Ballard, A.J.; Tintut, Y.; Demer, L.L. Role of the cholesterol biosynthetic pathway in osteoblastic differentiation of marrow stromal cells. J. Bone Miner. Res. 2002, 17, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Aghaloo, T.L.; Amantea, C.M.; Cowan, C.M.; Richardson, J.A.; Wu, B.M.; Parhami, F.; Tetradis, S. Oxysterols enhance osteoblast differentiation in vitro and bone healing in vivo. J. Orthop. Res. 2007, 25, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Guo, H.; Li, H. Cholesterol loading affects osteoblastic differentiation in mouse mesenchymal stem cells. Steroids 2013, 78, 426–433. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Sheng, Z.Y.; Tang, C.L.; Chen, L.; Pan, L.; Chen, J.Y. High cholesterol diet increases osteoporosis risk via inhibiting bone formation in rats. Acta Pharmacol. Sin. 2011, 32, 1498–1504. [Google Scholar] [CrossRef] [PubMed]
- Parhami, F.; Jackson, S.M.; Tintut, Y.; Le, V.; Balucan, J.P.; Territo, M.; Demer, L.L. Atherogenic diet and minimally oxidized low density lipoprotein inhibit osteogenic and promote adipogenic differentiation of marrow stromal cells. J. Bone Miner. Res. 1999, 14, 2067–2078. [Google Scholar] [CrossRef]
- Mangaraj, M.; Nanda, R.; Panda, S. Apolipoprotein A-I: A Molecule of Diverse Function. Indian. J. Clin. Biochem. 2016, 31, 253–259. [Google Scholar] [CrossRef]
- Blair, H.C.; Kalyvioti, E.; Papachristou, N.I.; Tourkova, I.L.; Syggelos, S.A.; Deligianni, D.; Orkoula, M.G.; Kontoyannis, C.G.; Karavia, E.A.; Kypreos, K.E.; et al. Apolipoprotein A-1 regulates osteoblast and lipoblast precursor cells in mice. Lab. Investig. 2016, 96, 763–772. [Google Scholar] [CrossRef]
- Shouhed, D.; Kha, H.T.; Richardson, J.A.; Amantea, C.M.; Hahn, T.J.; Parhami, F. Osteogenic oxysterols inhibit the adverse effects of oxidative stress on osteogenic differentiation of marrow stromal cells. J. Cell. Biochem. 2005, 95, 1276–1283. [Google Scholar] [CrossRef]
- Maziere, C.; Salle, V.; Gomila, C.; Maziere, J.C. Oxidized low density lipoprotein enhanced RANKL expression in human osteoblast-like cells. Involvement of ERK, NFkappaB and NFAT. Biochim. Biophys. Acta 2013, 1832, 1756–1764. [Google Scholar] [CrossRef]
- Maziere, C.; Savitsky, V.; Galmiche, A.; Gomila, C.; Massy, Z.; Maziere, J.C. Oxidized low density lipoprotein inhibits phosphate signaling and phosphate-induced mineralization in osteoblasts. Involvement of oxidative stress. Biochim. Biophys. Acta 2010, 1802, 1013–1019. [Google Scholar] [CrossRef]
- Harun, N.H.; Froemming, G.R.A.; Mohd Ismail, A.; Nawawi, H.; Mokhtar, S.S.; Abd Muid, S. Osteoblast Demineralization Induced by Oxidized High-Density Lipoprotein via the Inflammatory Pathway Is Suppressed by Adiponectin. Int. J. Mol. Sci. 2022, 23, 14616. [Google Scholar] [CrossRef]
- Pelton, K.; Krieder, J.; Joiner, D.; Freeman, M.R.; Goldstein, S.A.; Solomon, K.R. Hypercholesterolemia promotes an osteoporotic phenotype. Am. J. Pathol. 2012, 181, 928–936. [Google Scholar] [CrossRef]
- Demigne, C.; Bloch-Faure, M.; Picard, N.; Sabboh, H.; Besson, C.; Remesy, C.; Geoffroy, V.; Gaston, A.T.; Nicoletti, A.; Hagege, A.; et al. Mice chronically fed a westernized experimental diet as a model of obesity, metabolic syndrome and osteoporosis. Eur. J. Nutr. 2006, 45, 298–306. [Google Scholar] [CrossRef]
- Parhami, F.; Tintut, Y.; Beamer, W.G.; Gharavi, N.; Goodman, W.; Demer, L.L. Atherogenic high-fat diet reduces bone mineralization in mice. J. Bone Miner. Res. 2001, 16, 182–188. [Google Scholar] [CrossRef]
- Chen, X.; Wang, C.; Zhang, K.; Xie, Y.; Ji, X.; Huang, H.; Yu, X. Reduced femoral bone mass in both diet-induced and genetic hyperlipidemia mice. Bone 2016, 93, 104–112. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, Y.; Lin, J.; Qiu, X.; Chen, L.; Pan, X.; Lu, Y.; Zhang, J.; Wang, Y.; Li, D.; et al. Low-density lipoprotein receptor deficiency impaired mice osteoblastogenesis in vitro. Biosci. Trends 2018, 11, 658–666. [Google Scholar] [CrossRef]
- Martineau, C.; Martin-Falstrault, L.; Brissette, L.; Moreau, R. The atherogenic Scarb1 null mouse model shows a high bone mass phenotype. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E48–E57. [Google Scholar] [CrossRef]
- Isales, C.M.; Zaidi, M.; Blair, H.C. ACTH is a novel regulator of bone mass. Ann. N. Acad. Sci. 2010, 1192, 110–116. [Google Scholar] [CrossRef]
- Tourkova, I.L.; Dobrowolski, S.F.; Secunda, C.; Zaidi, M.; Papadimitriou-Olivgeri, I.; Papachristou, D.J.; Blair, H.C. The high-density lipoprotein receptor Scarb1 is required for normal bone differentiation in vivo and in vitro. Lab. Investig. 2019, 99, 1850–1860. [Google Scholar] [CrossRef]
- Alekos, N.S.; Moorer, M.C.; Riddle, R.C. Dual Effects of Lipid Metabolism on Osteoblast Function. Front. Endocrinol. 2020, 11, 578194. [Google Scholar] [CrossRef]
- Helderman, R.C.; Whitney, D.G.; Duta-Mare, M.; Akhmetshina, A.; Vujic, N.; Jayapalan, S.; Nyman, J.S.; Misra, B.B.; Rosen, C.J.; Czech, M.P.; et al. Loss of function of lysosomal acid lipase (LAL) profoundly impacts osteoblastogenesis and increases fracture risk in humans. Bone 2021, 148, 115946. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, H. Lysosomal Acid Lipase in Lipid Metabolism and Beyond. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Heur, M.; Duanmu, M.; Grabowski, G.A.; Hui, D.Y.; Witte, D.P.; Mishra, J. Lysosomal acid lipase-deficient mice: Depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. J. Lipid Res. 2001, 42, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Mangu, S.R.; Patel, K.; Sukhdeo, S.V.; Savitha, M.R.; Sharan, K. Maternal high-cholesterol diet negatively programs offspring bone development and downregulates hedgehog signaling in osteoblasts. J. Biol. Chem. 2022, 298, 102324. [Google Scholar] [CrossRef] [PubMed]
- DuSell, C.D.; Nelson, E.R.; Wang, X.; Abdo, J.; Modder, U.I.; Umetani, M.; Gesty-Palmer, D.; Javitt, N.B.; Khosla, S.; McDonnell, D.P. The endogenous selective estrogen receptor modulator 27-hydroxycholesterol is a negative regulator of bone homeostasis. Endocrinology 2010, 151, 3675–3685. [Google Scholar] [CrossRef]
- Nelson, E.R.; DuSell, C.D.; Wang, X.; Howe, M.K.; Evans, G.; Michalek, R.D.; Umetani, M.; Rathmell, J.C.; Khosla, S.; Gesty-Palmer, D.; et al. The oxysterol, 27-hydroxycholesterol, links cholesterol metabolism to bone homeostasis through its actions on the estrogen and liver X receptors. Endocrinology 2011, 152, 4691–4705. [Google Scholar] [CrossRef]
- Tarakida, A.; Iino, K.; Abe, K.; Taniguchi, R.; Higuchi, T.; Mizunuma, H.; Nakaji, S. Hypercholesterolemia accelerates bone loss in postmenopausal women. Climacteric 2011, 14, 105–111. [Google Scholar] [CrossRef]
- Go, J.H.; Song, Y.M.; Park, J.H.; Park, J.Y.; Choi, Y.H. Association between Serum Cholesterol Level and Bone Mineral Density at Lumbar Spine and Femur Neck in Postmenopausal Korean Women. Korean J. Fam. Med. 2012, 33, 166–173. [Google Scholar] [CrossRef]
- Tanko, L.B.; Bagger, Y.Z.; Nielsen, S.B.; Christiansen, C. Does serum cholesterol contribute to vertebral bone loss in postmenopausal women? Bone 2003, 32, 8–14. [Google Scholar] [CrossRef]
- Yamauchi, M.; Yamaguchi, T.; Nawata, K.; Tanaka, K.; Takaoka, S.; Sugimoto, T. Increased low-density lipoprotein cholesterol level is associated with non-vertebral fractures in postmenopausal women. Endocrine 2015, 48, 279–286. [Google Scholar] [CrossRef]
- Uyama, O.; Yoshimoto, Y.; Yamamoto, Y.; Kawai, A. Bone changes and carotid atherosclerosis in postmenopausal women. Stroke 1997, 28, 1730–1732. [Google Scholar] [CrossRef]
- Bagger, Y.Z.; Rasmussen, H.B.; Alexandersen, P.; Werge, T.; Christiansen, C.; Tanko, L.B.; PERF Study Group. Links between cardiovascular disease and osteoporosis in postmenopausal women: Serum lipids or atherosclerosis per se? Osteoporos. Int. 2007, 18, 505–512. [Google Scholar] [CrossRef]
- Majima, T.; Komatsu, Y.; Fukao, A.; Ninomiya, K.; Matsumura, T.; Nakao, K. Short-term effects of atorvastatin on bone turnover in male patients with hypercholesterolemia. Endocr. J. 2007, 54, 145–151. [Google Scholar] [CrossRef]
- Majima, T.; Shimatsu, A.; Komatsu, Y.; Satoh, N.; Fukao, A.; Ninomiya, K.; Matsumura, T.; Nakao, K. Short-term effects of pitavastatin on biochemical markers of bone turnover in patients with hypercholesterolemia. Intern. Med. 2007, 46, 1967–1973. [Google Scholar] [CrossRef]
- Yerges-Armstrong, L.M.; Shen, H.; Ryan, K.A.; Streeten, E.A.; Shuldiner, A.R.; Mitchell, B.D. Decreased bone mineral density in subjects carrying familial defective apolipoprotein B-100. J. Clin. Endocrinol. Metab. 2013, 98, E1999–E2005. [Google Scholar] [CrossRef]
- Awan, Z.; Alwaili, K.; Alshahrani, A.; Langsetmo, L.; Goltzman, D.; Genest, J. Calcium homeostasis and skeletal integrity in individuals with familial hypercholesterolemia and aortic calcification. Clin. Chem. 2010, 56, 1599–1607. [Google Scholar] [CrossRef]
- Hernandez, J.L.; Olmos, J.M.; Ramos, C.; Martinez, J.; de Juan, J.; Valero, C.; Nan, D.; Gonzalez-Macias, J. Serum lipids and bone metabolism in Spanish men: The Camargo cohort study. Endocr. J. 2010, 57, 51–60. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, G.; Zhang, Y.; Xu, G.; Yi, X.; Liang, J.; Zhao, C.; Liang, J.; Ma, C.; Ye, Y.; et al. Association Between Bone Mineral Density, Bone Turnover Markers, and Serum Cholesterol Levels in Type 2 Diabetes. Front. Endocrinol. 2018, 9, 646. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef]
- Bloch, K. The Biological Synthesis of Cholesterol. Science 1965, 150, 19–28. [Google Scholar] [CrossRef]
- Mundy, G.; Garrett, R.; Harris, S.; Chan, J.; Chen, D.; Rossini, G.; Boyce, B.; Zhao, M.; Gutierrez, G. Stimulation of bone formation in vitro and in rodents by statins. Science 1999, 286, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Mandal, C.C. High Cholesterol Deteriorates Bone Health: New Insights into Molecular Mechanisms. Front. Endocrinol. 2015, 6, 165. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Choudhury, N.; Mandal, C.C.; Choudhury, G.G. Statin-induced Ras activation integrates the phosphatidylinositol 3-kinase signal to Akt and MAPK for bone morphogenetic protein-2 expression in osteoblast differentiation. J. Biol. Chem. 2007, 282, 4983–4993. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Bertl, K.; Sun, H.; Liu, Z.H.; Andrukhov, O.; Rausch-Fan, X. Effect of simvastatin on the osteogenetic behavior of alveolar osteoblasts and periodontal ligament cells. Hum. Cell. 2012, 25, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Matsunuma, A.; Kurahashi, I.; Yanagawa, T.; Yoshida, H.; Horiuchi, N. Induction of osteoblast differentiation indices by statins in MC3T3-E1 cells. J. Cell. Biochem. 2004, 92, 458–471. [Google Scholar] [CrossRef]
- Tsubaki, M.; Satou, T.; Itoh, T.; Imano, M.; Yanae, M.; Kato, C.; Takagoshi, R.; Komai, M.; Nishida, S. Bisphosphonate- and statin-induced enhancement of OPG expression and inhibition of CD9, M-CSF, and RANKL expressions via inhibition of the Ras/MEK/ERK pathway and activation of p38MAPK in mouse bone marrow stromal cell line ST2. Mol. Cell. Endocrinol. 2012, 361, 219–231. [Google Scholar] [CrossRef]
- Ruan, F.; Zheng, Q.; Wang, J. Mechanisms of bone anabolism regulated by statins. Biosci. Rep. 2012, 32, 511–519. [Google Scholar] [CrossRef]
- Gong, Y.; Slee, R.B.; Fukai, N.; Rawadi, G.; Roman-Roman, S.; Reginato, A.M.; Wang, H.; Cundy, T.; Glorieux, F.H.; Lev, D.; et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001, 107, 513–523. [Google Scholar] [CrossRef]
- Lappalainen, S.; Saarinen, A.; Utriainen, P.; Voutilainen, R.; Jaaskelainen, J.; Makitie, O. LRP5 in premature adrenarche and in metabolic characteristics of prepubertal children. Clin. Endocrinol. 2009, 70, 725–731. [Google Scholar] [CrossRef]
- Suwazono, Y.; Kobayashi, E.; Uetani, M.; Miura, K.; Morikawa, Y.; Ishizaki, M.; Kido, T.; Nakagawa, H.; Nogawa, K. G-protein beta 3 subunit polymorphism C1429T and low-density lipoprotein receptor-related protein 5 polymorphism A1330V are risk factors for hypercholesterolemia in Japanese males--a prospective study over 5 years. Metabolism 2006, 55, 751–757. [Google Scholar] [CrossRef]
- Suwazono, Y.; Kobayashi, E.; Uetani, M.; Miura, K.; Morikawa, Y.; Ishizaki, M.; Kido, T.; Nakagawa, H.; Nogawa, K. Low-density lipoprotein receptor-related protein 5 variant Q89R is associated with hypertension in Japanese females. Blood Press. 2006, 15, 80–87. [Google Scholar] [CrossRef]
- Guo, Y.F.; Xiong, D.H.; Shen, H.; Zhao, L.J.; Xiao, P.; Guo, Y.; Wang, W.; Yang, T.L.; Recker, R.R.; Deng, H.W. Polymorphisms of the low-density lipoprotein receptor-related protein 5 (LRP5) gene are associated with obesity phenotypes in a large family-based association study. J. Med. Genet. 2006, 43, 798–803. [Google Scholar] [CrossRef]
- Fujino, T.; Asaba, H.; Kang, M.J.; Ikeda, Y.; Sone, H.; Takada, S.; Kim, D.H.; Ioka, R.X.; Ono, M.; Tomoyori, H.; et al. Low-density lipoprotein receptor-related protein 5 (LRP5) is essential for normal cholesterol metabolism and glucose-induced insulin secretion. Proc. Natl. Acad. Sci. USA 2003, 100, 229–234. [Google Scholar] [CrossRef]
- Little, R.D.; Carulli, J.P.; Del Mastro, R.G.; Dupuis, J.; Osborne, M.; Folz, C.; Manning, S.P.; Swain, P.M.; Zhao, S.C.; Eustace, B.; et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am. J. Hum. Genet. 2002, 70, 11–19. [Google Scholar] [CrossRef]
- Wang, B.; Wang, H.; Li, Y.; Song, L. Lipid metabolism within the bone micro-environment is closely associated with bone metabolism in physiological and pathophysiological stages. Lipids Health Dis. 2022, 21, 5. [Google Scholar] [CrossRef]
- Shen, G.; Ren, H.; Shang, Q.; Zhao, W.; Zhang, Z.; Yu, X.; Tang, K.; Tang, J.; Yang, Z.; Liang, D.; et al. Foxf1 knockdown promotes BMSC osteogenesis in part by activating the Wnt/beta-catenin signalling pathway and prevents ovariectomy-induced bone loss. EBioMedicine 2020, 52, 102626. [Google Scholar] [CrossRef]
- Foldi, J.; Chung, A.Y.; Xu, H.; Zhu, J.; Outtz, H.H.; Kitajewski, J.; Li, Y.; Hu, X.; Ivashkiv, L.B. Autoamplification of Notch signaling in macrophages by TLR-induced and RBP-J-dependent induction of Jagged1. J. Immunol. 2010, 185, 5023–5031. [Google Scholar] [CrossRef]
- Bjornson, C.R.; Cheung, T.H.; Liu, L.; Tripathi, P.V.; Steeper, K.M.; Rando, T.A. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 2012, 30, 232–242. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, D.; Guo, D.; Li, J.; Xu, S.; Wei, J.; Xie, J.; Zhou, X. Osteoblasts impair cholesterol synthesis in chondrocytes via Notch1 signalling. Cell. Prolif. 2021, 54, e13156. [Google Scholar] [CrossRef]
- Colombo, M.; Platonova, N.; Giannandrea, D.; Palano, M.T.; Basile, A.; Chiaramonte, R. Re-establishing Apoptosis Competence in Bone Associated Cancers via Communicative Reprogramming Induced Through Notch Signaling Inhibition. Front. Pharmacol. 2019, 10, 145. [Google Scholar] [CrossRef]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Dominguez, A.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The bone marrow microenvironment at single-cell resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Xiu, C.; Zhou, Q.; Ni, L.; Du, J.; Gong, T.; Li, M.; Saijilafu; Yang, H.; Chen, J. A dual role of cholesterol in osteogenic differentiation of bone marrow stromal cells. J. Cell. Physiol. 2019, 234, 2058–2066. [Google Scholar] [CrossRef] [PubMed]
- Golub, E.E. Role of matrix vesicles in biomineralization. Biochim. Biophys. Acta 2009, 1790, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Millan, J.L. The role of phosphatases in the initiation of skeletal mineralization. Calcif. Tissue Int. 2013, 93, 299–306. [Google Scholar] [CrossRef]
- Favarin, B.Z.; Andrade, M.A.R.; Bolean, M.; Simao, A.M.S.; Ramos, A.P.; Hoylaerts, M.F.; Millan, J.L.; Ciancaglini, P. Effect of the presence of cholesterol in the interfacial microenvironment on the modulation of the alkaline phosphatase activity during in vitro mineralization. Colloids Surf. B Biointerfaces 2017, 155, 466–476. [Google Scholar] [CrossRef]
- Vimalraj, S. Alkaline phosphatase: Structure, expression and its function in bone mineralization. Gene 2020, 754, 144855. [Google Scholar] [CrossRef]
- Luu, W.; Sharpe, L.J.; Gelissen, I.C.; Brown, A.J. The role of signalling in cellular cholesterol homeostasis. IUBMB Life 2013, 65, 675–684. [Google Scholar] [CrossRef]
- Nelson, D.L.; Hoskins, A.A.; Cox, M.M.; Lehninger, A.L. Lehninger Principles of Biochemistry, 8th ed.; Macmillan Learning: Austin, TX, USA, 2021. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Research Object | Treatment/Diet | Cholesterol/Cholesterol Derivatives Concentration | Effect | Reference |
|---|---|---|---|---|
| M2-10B4 (mouse stromal cell line) | 22(S)-hydroxycholesterol 20(S)-hydroxycholesterol (SS) | 5 mM | ↑ALP activity ↑mineralization ↑OCN mRNA | [43] |
| MSCs (bone marrow- derived mesenchymal stem cells) | Chol:MbCD (‘‘water-soluble cholesterol’’ containing 30 mg of cholesterol/g solid) | 5, 10 and 15 μg/mL | ↑differentiation ↑ALP activity ↑mineralized nodules | [44] |
| MC3T3-E1 (mouse cell line of immature osteoblasts) | Cholesterol | 0, 12.5, 25, and 50 μg/mL | ↓proliferation ↓differentiation ↑oxidative injury | [45] |
| M2–10B4 | Oxidized low-density lipoprotein (MM-LDL) | 150 µg/mL | ↓osteogenic differentiation ↑adipogenic differentiation | [46] |
| M2-10B4, Primary mouse bone marrow stromal cells | Xanthine/xanthine oxidase (XXO) minimally oxidized LDL (MM-LDL) Osteogenic oxysterol combination 22(S)- and 20(S)-hydroxycholesterol | 50 mM/40 mU/mL 200 mg/mL 0.1–5 µM | ↓markers of osteogenic differentiation blocked and reversed the inhibition of osteogenic differentiation | [49] |
| UMR106 (rat osteoblast-like cell line) | Oxidized LDL (oxLDL) | 10–50 μg protein/mL | ↓mineralization | [51] |
| MG63 (human osteosarcoma cell line) | Oxidized LDL (oxLDL) | 10–50 μg/mL | ↑cell-associated and extracellular RANKL levels | [50] |
| HOBs (primary human osteoblast cells) | Oxidized HDL (oxHDL) oxHDL with adiponectin | 100 μg/mL protein 100 μg/mL; 5, 10, and 15 μg/mL | ↓mineralization, ↓calcium incorporation. ↑expression of mineralization markers ↓inflammatory markers | [52] |
| Rat | Poly (lactic-co-glycolic acid) (PLGA) scaffolds alone or oxysterol cocktail | 140 ng (low dose) 1400 ng (high dose) | slight bone healing ↑bone formation | [43] |
| Rat | High-cholesterol diet | 77% normal diet food, 3% cholesterol and 20% lard | ↓femur BMD ↓osteocalcin ↑carboxy-terminal collagen crosslinks | [45] |
| C57BL/6 and C.B-17/Icr-SCID/Sed-Prkdcscid male mice | High-fat/high-cholesterol (HFHC) diet | 1.25% cholesterol | ↓cortical and trabecular bone in the femurs and vertebrae ↓bone mineral density (BMD) | [53] |
| OF1 female mice | Westerntype diet | 1.1 mg cholesterol/g diet | ↓BMD | [54] |
| C57BL/6 and C3H/HeJ male mice | High-fat (atherogenic) diet | 1.25% cholesterol | ↓femoral and vertebral mineral content ↓BMD | [55] |
| C57BL6/J and Swiss Albino mice | High-cholesterol (HC) diet | 0.5% cholesterol | ↓osteoblast cell activity ↑osteoclast cell population delayed skeletal ossification | [65] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhmetshina, A.; Kratky, D.; Rendina-Ruedy, E. Influence of Cholesterol on the Regulation of Osteoblast Function. Metabolites 2023, 13, 578. https://doi.org/10.3390/metabo13040578
Akhmetshina A, Kratky D, Rendina-Ruedy E. Influence of Cholesterol on the Regulation of Osteoblast Function. Metabolites. 2023; 13(4):578. https://doi.org/10.3390/metabo13040578
Chicago/Turabian StyleAkhmetshina, Alena, Dagmar Kratky, and Elizabeth Rendina-Ruedy. 2023. "Influence of Cholesterol on the Regulation of Osteoblast Function" Metabolites 13, no. 4: 578. https://doi.org/10.3390/metabo13040578
APA StyleAkhmetshina, A., Kratky, D., & Rendina-Ruedy, E. (2023). Influence of Cholesterol on the Regulation of Osteoblast Function. Metabolites, 13(4), 578. https://doi.org/10.3390/metabo13040578