
Hepatic Insulin Resistance Model in the Male Wistar Rat Using Exogenous Insulin Glargine Administration
 , , , , and
, , , , and
Abstract
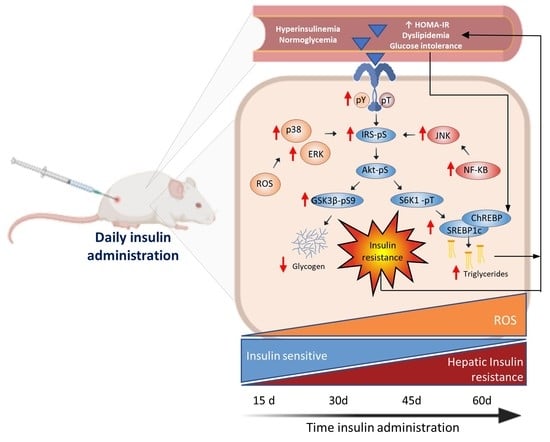
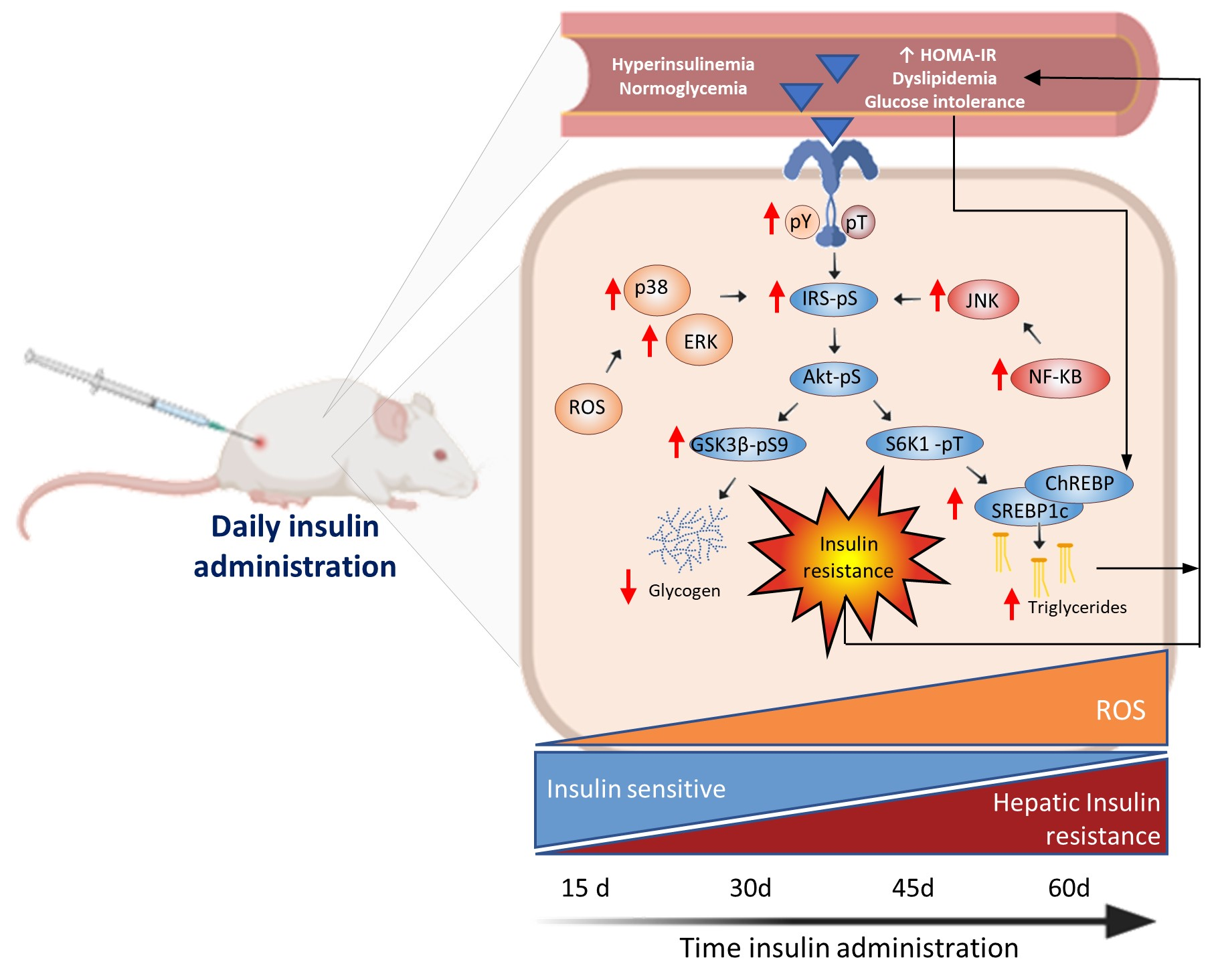
1. Introduction
2. Materials and Methods
2.1. Animals and Treatment
2.2. Zoometry
2.3. Oral Glucose Tolerance Test, Insulin Response, and Insulin Resistance Analyses
2.4. Biochemical Assays
2.5. Tissue for Biochemical, Immunoassay, and Histological Tests
2.6. Biochemical Assays
2.7. Redox Balance Assays
2.8. Immunoassays
2.9. Histological Assays
2.10. Statistical Analysis
3. Results
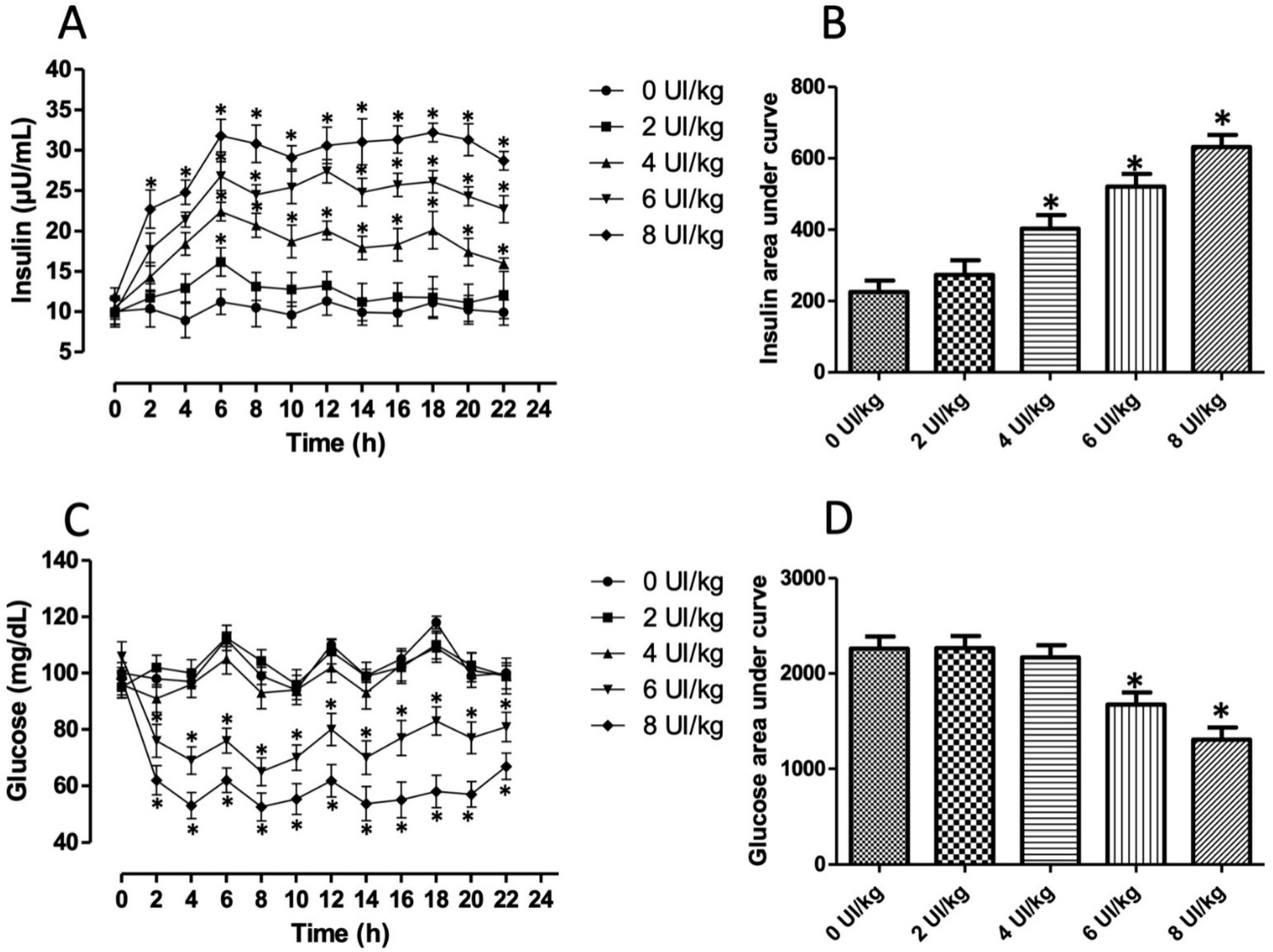
3.1. Hyperinsulinemia-Euglycemia Model
3.2. Insulin Resistance Model
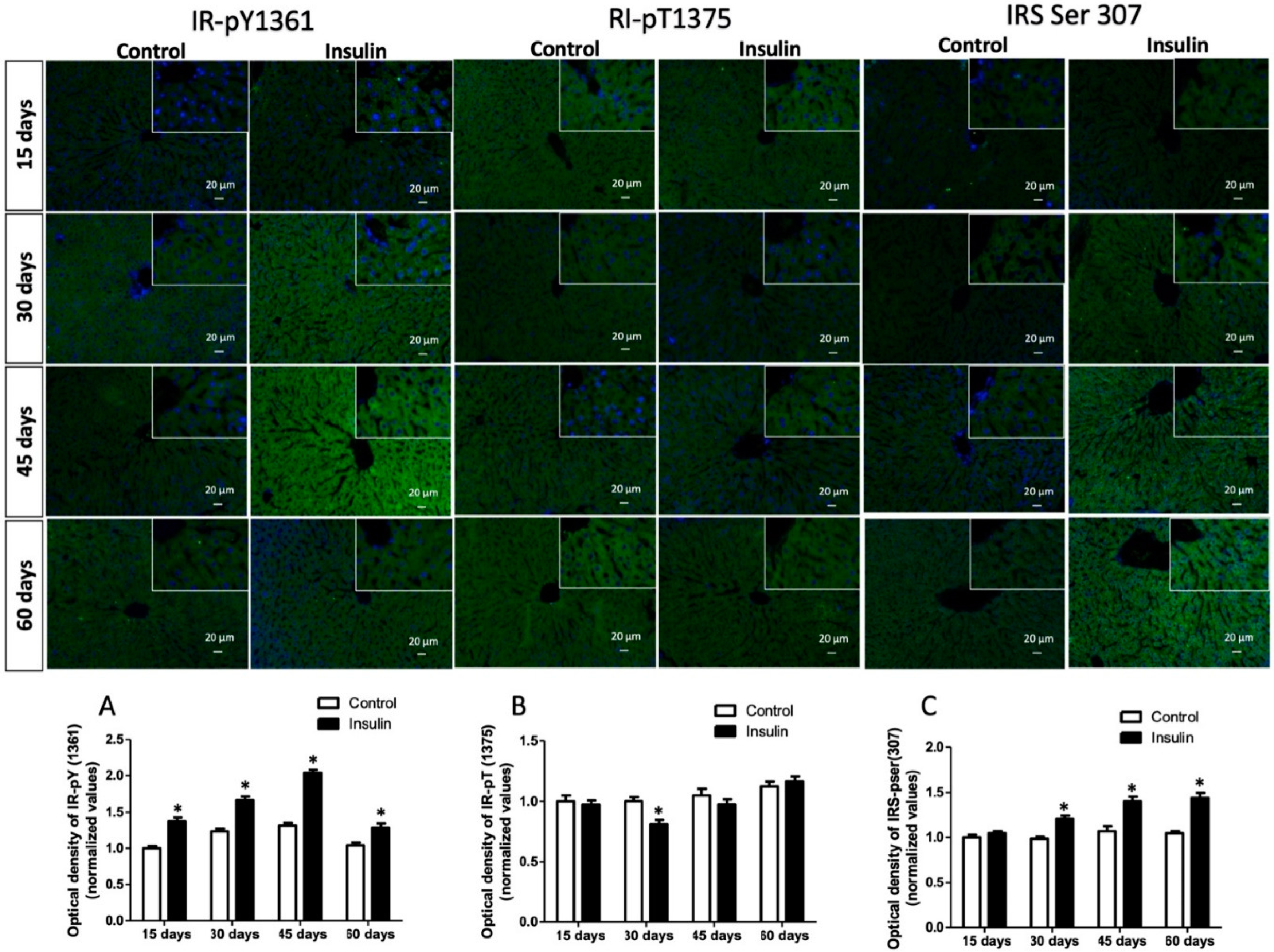
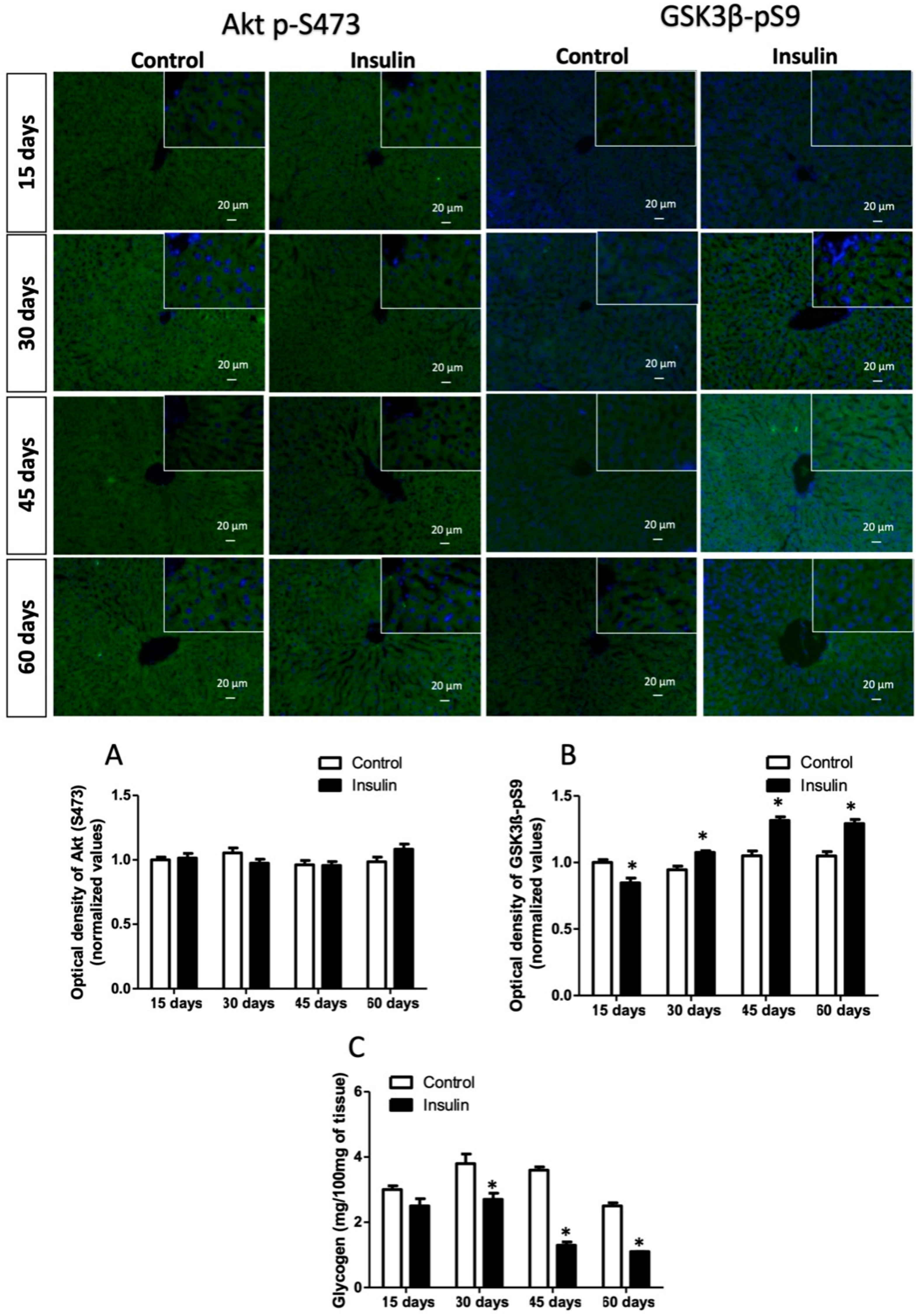
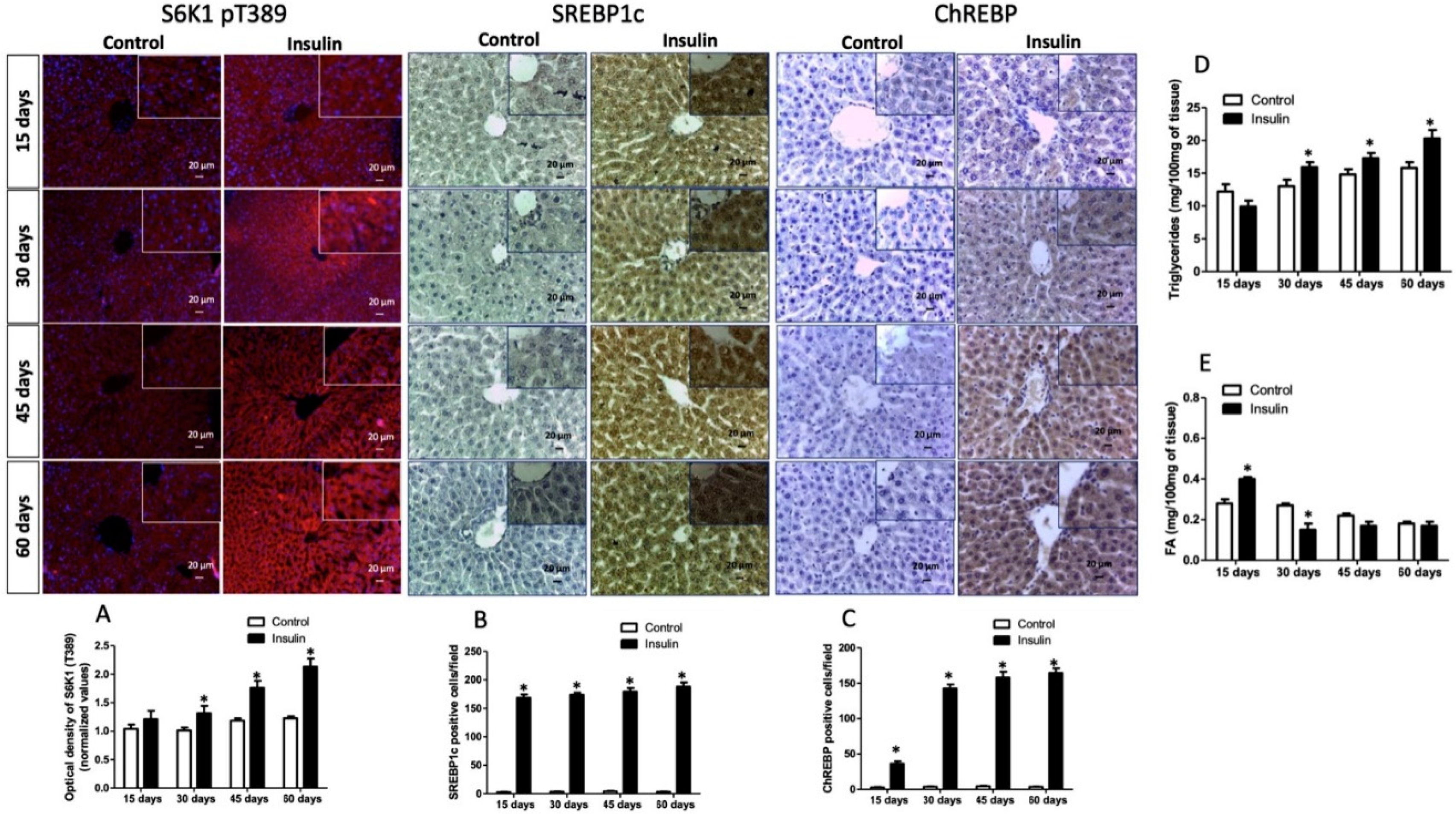
3.3. Glargine Administration Impairs Hepatic Insulin Metabolic Signaling
3.4. Effect of Glargine Administration on Serum Lipid Profile
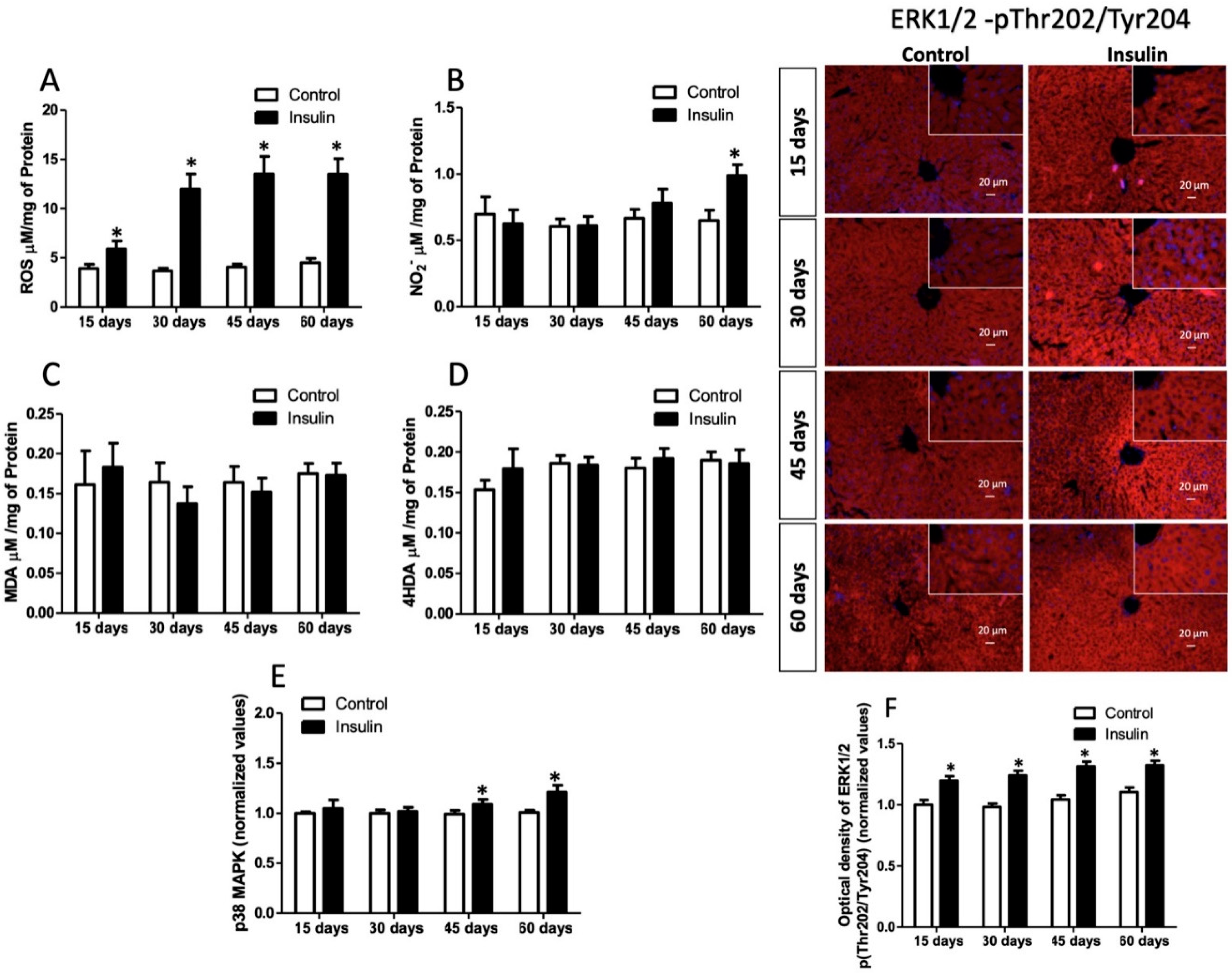
3.5. Effect of Insulin Glargine Administration on Hepatic Redox Balance and MAPK Response
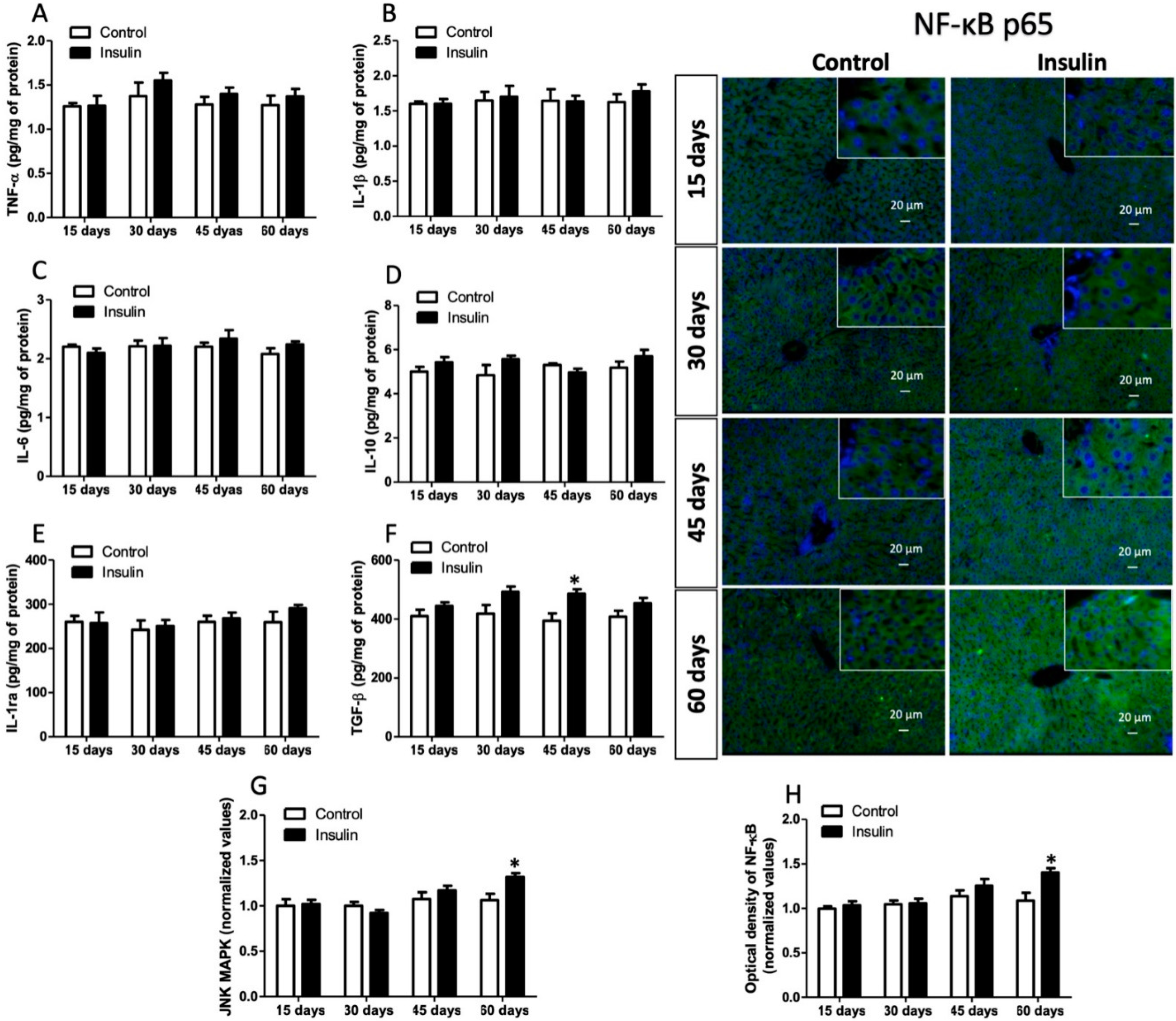
3.6. Insulin Glargine Administration on the Hepatic Inflammation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res. Clin. Pr. 2022, 183, 109119. [Google Scholar] [CrossRef]
- Ebrahimoghli, R.; Janati, A.; Sadeghi-Bazargani, H.; Hamishehkar, H.; Khalili-Azimi, A. Incremental Healthcare Resource Utilization and Expenditures Associated with Cardiovascular Diseases in Patients with Diabetes: A Cross-Sectional Study. Iran. J. Med. Sci. 2022, 47, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Park, S.Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15. [Google Scholar] [CrossRef]
- Sarmiento-Ortega, V.E.; Moroni-González, D.; Diaz, A.; Brambila, E.; Treviño, S. ROS and ERK Pathway Mechanistic Approach on Hepatic Insulin Resistance after Chronic Oral Exposure to Cadmium NOAEL Dose. Biol. Trace Elem. Res. 2022, 8, 1–16. [Google Scholar] [CrossRef]
- Sarmiento-Ortega, V.E.; Moroni-González, D.; Díaz, A.; Eduardo, B.; Samuel, T. Oral Subacute Exposure to Cadmium LOAEL Dose Induces Insulin Resistance and Impairment of the Hormonal and Metabolic Liver-Adipose Axis in Wistar Rats. Biol. Trace Elem. Res. 2021, 10, 4370–4384. [Google Scholar] [CrossRef]
- Treviño, S.; Díaz, A.; González-López, G.; Guevara, J. Differential Biochemical-Inflammatory Patterns in the Astrocyte-Neuron Axis of the Hippocampus and Frontal Cortex in Wistar Rats with Metabolic Syndrome Induced by High Fat or Carbohydrate Diets. J. Chem. Neuroanat. 2022, 126, 102186. [Google Scholar] [CrossRef]
- Da Silva Rosa, S.C.; Nayak, N.; Caymo, A.M.; Gordon, J.W. Mechanisms of Muscle Insulin Resistance and the Cross-Talk with Liver and Adipose Tissue. Physiol. Rep. 2020, 8, e14607. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Treviño, S.; Diaz, A. Vanadium and Insulin: Partners in Metabolic Regulation. J. Inorg. Biochem. 2020, 208, 111094. [Google Scholar] [CrossRef] [PubMed]
- Treviño, S.; Díaz, A.; Sánchez-Lara, E.; Sanchez-Gaytan, B.L.; Perez-Aguilar, J.M.; González-Vergara, E. Vanadium in Biological Action: Chemical, Pharmacological Aspects, and Metabolic Implications in Diabetes Mellitus. Biol. Trace Elem. Res. 2019, 188, 68–98. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Kahn, C.R. Insulin Signalling and the Regulation of Glucose and Lipid Metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of Hepatic Glucose Metabolism in Health and Disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef]
- Treviño, S.; Cortezano-Esteban, S.; Hernández-Fragoso, H.; Díaz, A.; Vázquez-Roque, R.; Enrique Sarmiento-Ortega, V.; Moroni-González, D.; Pelayo, R.; Brambila, E. Clinical Monitored in Subjects Metabolically Healthy and Unhealthy before and during a SARS-CoV-2 Infection- A Cross-Sectional Study in Mexican Population. Cytokine 2022, 153, 155868. [Google Scholar] [CrossRef]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Type 2 Diabetes—An Autoinflammatory Disease Driven by Metabolic Stress. Biochim. Biophys. Acta. Mol. Basis. Dis. 2018, 1864, 3805–3823. [Google Scholar] [CrossRef]
- Aguirre, V.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The C-Jun NH(2)-Terminal Kinase Promotes Insulin Resistance during Association with Insulin Receptor Substrate-1 and Phosphorylation of Ser(307). J. Biol. Chem. 2000, 275, 9047–9054. [Google Scholar] [CrossRef]
- Wei, Y.; Pagliassotti, M.J. Hepatospecific Effects of Fructose on C-Jun NH2-Terminal Kinase: Implications for Hepatic Insulin Resistance. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E926–E933. [Google Scholar] [CrossRef] [PubMed]
- Mastrototaro, L.; Roden, M. Insulin Resistance and Insulin Sensitizing Agents. Metabolism 2021, 125, 154892. [Google Scholar] [CrossRef]
- Krako Jakovljevic, N.; Pavlovic, K.; Zujovic, T.; Kravic-Stevovic, T.; Jotic, A.; Markovic, I.; Lalic, N.M. In Vitro Models of Insulin Resistance: Mitochondrial Coupling Is Differently Affected in Liver and Muscle Cells. Mitochondrion 2021, 61, 165–173. [Google Scholar] [CrossRef]
- Kleinert, M.; Clemmensen, C.; Hofmann, S.M.; Moore, M.C.; Renner, S.; Woods, S.C.; Huypens, P.; Beckers, J.; de Angelis, M.H.; Schürmann, A.; et al. Animal Models of Obesity and Diabetes Mellitus. Nat. Rev. Endocrinol. 2018, 14, 140–162. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento-Ortega, V.; Brambila, E.; Flores-Hernández, J.; Díaz, A.; Peña-Rosas, U.; Moroni-González, D.; Aburto-Luna, V.; Treviño, S. The NOAEL Metformin Dose Is Ineffective against Metabolic Disruption Induced by Chronic Cadmium Exposure in Wistar Rats. Toxics 2018, 6, 55. [Google Scholar] [CrossRef]
- Treviño, S.; Waalkes, M.P.; Flores Hernández, J.A.; León-Chavez, B.A.; Aguilar-Alonso, P.; Brambila, E. Chronic Cadmium Exposure in Rats Produces Pancreatic Impairment and Insulin Resistance in Multiple Peripheral Tissues. Arch. Biochem. Biophys. 2015, 583, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.W.; Keirs, R.W.; Peebles, E.D.; Gerard, P.D. Methodologies of Tissue Preservation and Analysis of the Glycogen Content of the Broiler Chick Liver. Poult. Sci. 2007, 86, 2653–2665. [Google Scholar] [CrossRef]
- Schonten, J.A.; Popp-Snijders, C. Determination of Free Fatty Acids: A Comparative Study of the Enzymatic Versus the Gas Chromatographie and the Colorimetric Method. Clin. Chem. Lab. Med. 1983, 21, 823–828. [Google Scholar] [CrossRef]
- Sedmak, J.J.; Grossberg, S.E. A Rapid, Sensitive, and Versatile Assay for Protein Using Coomassie Brilliant Blue G250. Anal. Biochem. 1977, 79, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Eaton, D.L.; George Cherian, M. Determination of Metallothionein in Tissues by Cadmium-Hemoglobin Affinity Assay. Methods Enzym. 1991, 205, 83–88. [Google Scholar] [CrossRef]
- Flohé, L.; Günzler, W.A. Assays of Glutathione Peroxidase. Methods Enzym. 1984, 105, 114–120. [Google Scholar] [CrossRef]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-Transferases. The First Enzymatic Step in Mercapturic Acid Formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [CrossRef]
- Smith, I.K.; Vierheller, T.L.; Thorne, C.A. Assay of Glutathione Reductase in Crude Tissue Homogenates Using 5,5′-Dithiobis(2-Nitrobenzoic Acid). Anal. Biochem. 1988, 175, 408–413. [Google Scholar] [CrossRef]
- Rosenstock, J.; Fonseca, V.; Schinzel, S.; Dain, M.P.; Mullins, P.; Riddle, M. Reduced Risk of Hypoglycemia with Once-Daily Glargine versus Twice-Daily NPH and Number Needed to Harm with NPH to Demonstrate the Risk of One Additional Hypoglycemic Event in Type 2 Diabetes: Evidence from a Long-Term Controlled Trial. J. Diabetes Complicat. 2014, 28, 742–749. [Google Scholar] [CrossRef]
- Juan, C.C.; Fang, V.S.; Kwok, C.F.; Perng, J.C.; Chou, Y.C.; Ho, L.T. Exogenous Hyperinsulinemia Causes Insulin Resistance, Hyperendothelinemia, and Subsequent Hypertension in Rats. Metabolism 1999, 48, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Dal, S.; Jeandidier, N.; Schaschkow, A.; Spizzo, A.H.; Seyfritz, E.; Sookhareea, C.; Bietiger, W.; Péronet, C.; Moreau, F.; Pinget, M.; et al. Portal or Subcutaneous Insulin Infusion: Efficacy and Impact on Liver Inflammation. Fundam. Clin. Pharm. 2015, 29, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Treviño, S.; González-Vergara, E. Metformin-Decavanadate Treatment Ameliorates Hyperglycemia and Redox Balance of the Liver and Muscle in a Rat Model of Alloxan-Induced Diabetes. New J. Chem. 2019, 43, 17850–17862. [Google Scholar] [CrossRef]
- Wu, X.; Chen, K.; Williams, K.J. The Role of Pathway-Selective Insulin Resistance and Responsiveness in Diabetic Dyslipoproteinemia. Curr. Opin. Lipidol. 2012, 23, 334–344. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Röhling, M.; Martin, S. Insulin: Too Much of a Good Thing Is Bad. BMC Med. 2020, 18, 224. [Google Scholar] [CrossRef]
- Nozaki, Y.; Petersen, M.C.; Zhang, D.; Vatner, D.F.; Perry, R.J.; Abulizi, A.; Haedersdal, S.; Zhang, X.M.; Butrico, G.M.; Samuel, V.T.; et al. Metabolic Control Analysis of Hepatic Glycogen Synthesis in Vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 8166–8176. [Google Scholar] [CrossRef]
- Bergman, R.N.; Iyer, M.S. Indirect Regulation of Endogenous Glucose Production by Insulin: The Single Gateway Hypothesis Revisited. Diabetes 2017, 66, 1742–1747. [Google Scholar] [CrossRef]
- Rines, A.K.; Sharabi, K.; Tavares, C.D.J.; Puigserver, P. Targeting Hepatic Glucose Metabolism in the Treatment of Type 2 Diabetes. Nat. Rev. Drug Discov. 2016, 15, 786–804. [Google Scholar] [CrossRef]
- Jiang, S.; Young, J.L.; Wang, K.; Qian, Y.; Cai, L. Diabetic-Induced Alterations in Hepatic Glucose and Lipid Metabolism: The Role of Type 1 and Type 2 Diabetes Mellitus (Review). Mol. Med. Rep. 2020, 22, 603–611. [Google Scholar] [CrossRef]
- Woerle, H.J.; Szoke, E.; Meyer, C.; Dostou, J.M.; Wittlin, S.D.; Gosmanov, N.R.; Welle, S.L.; Gerich, J.E. Mechanisms for Abnormal Postprandial Glucose Metabolism in Ty.ype 2 Diabetes. Am. J. Physiol. Endocrinol. Metab. 2006, 290, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Brown, M.S.; Goldstein, J.L. Bifurcation of Insulin Signaling Pathway in Rat Liver: MTORC1 Required for Stimulation of Lipogenesis, but Not Inhibition of Gluconeogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3441–3446. [Google Scholar] [CrossRef]
- Shimano, H.; Sato, R. SREBP-Regulated Lipid Metabolism: Convergent Physiology—Divergent Pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Owen, J.L.; Zhang, Y.; Bae, S.H.; Farooqi, M.S.; Liang, G.; Hammer, R.E.; Goldstein, J.L.; Brown, M.S. Insulin Stimulation of SREBP-1c Processing in Transgenic Rat Hepatocytes Requires P70 S6-Kinase. Proc. Natl. Acad. Sci. USA 2012, 109, 16184–16189. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Takao, K.; Yabe, D. ChREBP-Mediated Regulation of Lipid Metabolism: Involvement of the Gut Microbiota, Liver, and Adipose Tissue. Front. Endocrinol. 2020, 11, 587189. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Tsuchida, H.; Kato, T.; Niwa, H.; Horikawa, Y.; Takeda, J.; Iizuka, K. Fat and Carbohydrate in Western Diet Contribute Differently to Hepatic Lipid Accumulation. Biochem. Biophys. Res. Commun. 2015, 461, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Badmus, O.O.; Hillhouse, S.A.; Anderson, C.D.; Hinds, T.D.; Stec, D.E. Molecular Mechanisms of Metabolic Associated Fatty Liver Disease (MAFLD): Functional Analysis of Lipid Metabolism Pathways. Clin. Sci. 2022, 136, 1347–1366. [Google Scholar] [CrossRef]
- Sakiyama, H.; Li, L.; Inoue, M.; Eguchi, H.; Yoshihara, D.; Fujiwara, N.; Suzuki, K. ChREBP Deficiency Prevents High Sucrose Diet-Induced Obesity through Reducing Sucrase Expression. J. Clin. Biochem. Nutr. 2022, 71, 221–228. [Google Scholar] [CrossRef]
- Dentin, R.; Benhamed, F.; Hainault, I.; Fauveau, V.; Foufelle, F.; Dyck, J.R.B.; Girard, J.; Postic, C. Liver-Specific Inhibition of ChREBP Improves Hepatic Steatosis and Insulin Resistance in Ob/Ob Mice. Diabetes 2006, 55, 2159–2170. [Google Scholar] [CrossRef]
- Watt, M.J.; Miotto, P.M.; de Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393. [Google Scholar] [CrossRef] [PubMed]
- Tanase, D.M.; Gosav, E.M.; Costea, C.F.; Ciocoiu, M.; Lacatusu, C.M.; Maranduca, M.A.; Ouatu, A.; Floria, M. The Intricate Relationship between Type 2 Diabetes Mellitus (T2DM), Insulin Resistance (IR), and Nonalcoholic Fatty Liver Disease (NAFLD). J. Diabetes Res. 2020, 2020, 3920196. [Google Scholar] [CrossRef]
- Haas, M.E.; Attie, A.D.; Biddinger, S.B. The Regulation of ApoB Metabolism by Insulin. Trends Endocrinol. Metab. 2013, 24, 391–397. [Google Scholar] [CrossRef]
- Heeren, J.; Scheja, L. Metabolic-Associated Fatty Liver Disease and Lipoprotein Metabolism. Mol. Metab. 2021, 50, 101238. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Taskinen, M.R.; Björnson, E.; Packard, C.J. Metabolism of Triglyceride-Rich Lipoproteins in Health and Dyslipidaemia. Nat. Rev. Cardiol. 2022, 19, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Adiels, M.; Taskinen, M.R.; Packard, C.; Caslake, M.J.; Soro-Paavonen, A.; Westerbacka, J.; Vehkavaara, S.; Häkkinen, A.; Olofsson, S.O.; Yki-Järvinen, H.; et al. Overproduction of Large VLDL Particles Is Driven by Increased Liver Fat Content in Man. Diabetologia 2006, 49, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Sanders, F.W.B.; Griffin, J.L. De Novo Lipogenesis in the Liver in Health and Disease: More than Just a Shunting Yard for Glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento-Ortega, V.E.; Treviño, S.; Flores-Hernández, J.Á.; Aguilar-Alonso, P.; Moroni-González, D.; Aburto-Luna, V.; Diaz, A.; Brambila, E. Changes on Serum and Hepatic Lipidome after a Chronic Cadmium Exposure in Wistar Rats. Arch. Biochem. Biophys. 2017, 635, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Treviño, S.; Velázquez-Vázquez, D.; Sánchez-Lara, E.; Diaz-Fonseca, A.; Flores-Hernandez, J.Á.; Pérez-Benítez, A.; Brambila-Colombres, E.; González-Vergara, E. Metforminium Decavanadate as a Potential Metallopharmaceutical Drug for the Treatment of Diabetes Mellitus. Oxid. Med. Cell Longev. 2016, 2016, 6058705. [Google Scholar] [CrossRef] [PubMed]
- Perla, F.M.; Prelati, M.; Lavorato, M.; Visicchio, D.; Anania, C. The Role of Lipid and Lipoprotein Metabolism in Non-Alcoholic Fatty Liver Disease. Children 2017, 4, 46. [Google Scholar] [CrossRef]
- Doege, H.; Grimm, D.; Falcon, A.; Tsang, B.; Storm, T.A.; Xu, H.; Ortegon, A.M.; Kazantzis, M.; Kay, M.A.; Stahl, A. Silencing of Hepatic Fatty Acid Transporter Protein 5 In Vivo Reverses Diet-Induced Non-Alcoholic Fatty Liver Disease and Improves Hyperglycemia. J. Biol. Chem. 2008, 283, 22186–22192. [Google Scholar] [CrossRef]
- Young, E.N.; Dogan, M.; Watkins, C.; Bajwa, A.; Eason, J.D.; Kuscu, C.; Kuscu, C. A Review of Defatting Strategies for Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 1805. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa-Nagata, N.; Takamura, T.; Ando, H.; Nakamura, S.; Kurita, S.; Misu, H.; Ota, T.; Yokoyama, M.; Honda, M.; Miyamoto, K.-i.; et al. Increased Oxidative Stress Precedes the Onset of High-Fat Diet-Induced Insulin Resistance and Obesity. Metabolism 2008, 57, 1071–1077. [Google Scholar] [CrossRef]
- Avram, V.F.; Merce, A.P.; Hâncu, I.M.; Bătrân, A.D.; Kennedy, G.; Rosca, M.G.; Muntean, D.M. Impairment of Mitochondrial Respiration in Metabolic Diseases: An Overview. Int. J. Mol. Sci. 2022, 23, 8852. [Google Scholar] [CrossRef] [PubMed]
- Ota, T.; Gayet, C.; Ginsberg, H.N. Inhibition of Apolipoprotein B100 Secretion by Lipid-Induced Hepatic Endoplasmic Reticulum Stress in Rodents. J. Clin. Invest. 2008, 118, 316–332. [Google Scholar] [CrossRef] [PubMed]
- Caviglia, J.M.; Gayet, C.; Ota, T.; Hernandez-Ono, A.; Conlon, D.M.; Jiang, H.; Fisher, E.A.; Ginsberg, H.N. Different Fatty Acids Inhibit ApoB100 Secretion by Different Pathways: Unique Roles for ER Stress, Ceramide, and Autophagy. J. Lipid Res. 2011, 52, 1636–1651. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.H.; Han, C.Y. Signaling Nodes Associated with Endoplasmic Reticulum Stress during NAFLD Progression. Biomolecules 2021, 11, 242. [Google Scholar] [CrossRef] [PubMed]
- Syafril, S.; Lindarto, D.; Lelo, A.; Sembiring, R.J.; Saragih, A. Correlations between Insulin Receptor Substrate-1 with Phosphoinositide 3-Kinase and P38 Mitogen-Activated Protein Kinase Levels after Treatment of Diabetic Rats with Puguntano (Curanga Fel-Terrae [Merr.]) Leaf Extract. Open Access Maced. J. Med. Sci. 2019, 7, 1247–1251. [Google Scholar] [CrossRef]
- Hornberg, J.J.; Binder, B.; Bruggeman, F.J.; Schoeberl, B.; Heinrich, R.; Westerhoff, H.V. Control of MAPK Signalling: From Complexity to What Really Matters. Oncogene 2005, 24, 5533–5542. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Zhu, W.; Wang, S.; Xie, C.; Li, X.; Wu, J.; Li, Y.; Chen, Y.; Wang, X.; Meng, Y.; et al. P53 Modulates Hepatic Insulin Sensitivity through NF-ΚB and P38/ERK MAPK Pathways. Biochem. Biophys. Res. Commun. 2018, 495, 2139–2144. [Google Scholar] [CrossRef] [PubMed]
- Kurutas, E.B. The Importance of Antioxidants Which Play the Role in Cellular Response against Oxidative/Nitrosative Stress: Current State. Nutr. J. 2016, 15, 1–22. [Google Scholar] [CrossRef]
- Yang, B.; Xu, B.; Zhao, H.; Wang, Y.B.; Zhang, J.; Li, C.W.; Wu, Q.; Cao, Y.K.; Li, Y.; Cao, F. Dioscin Protects against Coronary Heart Disease by Reducing Oxidative Stress and Inflammation via Sirt1/Nrf2 and P38 MAPK Pathways. Mol. Med. Rep. 2018, 18, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and Mitochondria. Front. Pharm. 2014, 5, 151. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Fernández-Galilea, M.; Martínez-Fernández, L.; González-Muniesa, P.; Pérez-Chávez, A.; Martínez, J.A.; Moreno-Aliaga, M.J. Oxidative Stress and Non-Alcoholic Fatty Liver Disease: Effects of Omega-3 Fatty Acid Supplementation. Nutrients 2019, 11, 872. [Google Scholar] [CrossRef]
- Romá-Mateo, C.; Seco-Cervera, M.; Ibáñez-Cabellos, J.S.; Pérez, G.; Berenguer-Pascual, E.; Rodríguez, L.R.; García-Giménez, J.L. Oxidative Stress and the Epigenetics of Cell Senescence: Insights from Progeroid Syndromes. Curr. Pharm. Des. 2019, 24, 4755–4770. [Google Scholar] [CrossRef]
- Pomatto, L.C.D.; Davies, K.J.A. Adaptive Homeostasis and the Free Radical Theory of Ageing. Free Radic. Biol. Med. 2018, 124, 420–430. [Google Scholar] [CrossRef]
- Huh, J.Y.; Saltiel, A.R. Roles of IκB Kinases and TANK-Binding Kinase 1 in Hepatic Lipid Metabolism and Nonalcoholic Fatty Liver Disease. Exp. Mol. Med. 2021, 53, 1697–1705. [Google Scholar] [CrossRef]
- Brenachot, X.; Ramadori, G.; Ioris, R.M.; Veyrat-Durebex, C.; Altirriba, J.; Aras, E.; Ljubicic, S.; Kohno, D.; Fabbiano, S.; Clement, S.; et al. Hepatic Protein Tyrosine Phosphatase Receptor Gamma Links Obesity-Induced Inflammation to Insulin Resistance. Nat. Commun. 2017, 8, 1820. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, F.T.; Luedde, T.; Singer, S.; Schmidt-Supprian, M.; Baumgartl, J.; Schirmacher, P.; Pasparakis, M.; Brüning, J.C. Hepatic NF-Kappa B Essential Modulator Deficiency Prevents Obesity-Induced Insulin Resistance but Synergizes with High-Fat Feeding in Tumorigenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 1297–1302. [Google Scholar] [CrossRef]
- Tang, T.; Zhang, J.; Yin, J.; Staszkiewicz, J.; Gawronska-Kozak, B.; Jung, D.Y.; Ko, H.J.; Ong, H.; Kim, J.K.; Mynatt, R.; et al. Uncoupling of Inflammation and Insulin Resistance by NF-KappaB in Transgenic Mice through Elevated Energy Expenditure. J. Biol. Chem. 2010, 285, 4637–4644. [Google Scholar] [CrossRef] [PubMed]
- Gasmi, A.; Piscopo, S.; Menzel, A.; Noor, S. A Review on Metabolic Paradoxes and Their Impact on Metabolism. Arch. Razi Inst. 2022, 77, 929–941. [Google Scholar] [CrossRef]
- Glintborg, D.; Højlund, K.; Andersen, N.R.; Hansen, B.F.; Beck-Nielsen, H.; Wojtaszewski, J.F.P. Impaired Insulin Activation and Dephosphorylation of Glycogen Synthase in Skeletal Muscle of Women with Polycystic Ovary Syndrome Is Reversed by Pioglitazone Treatment. J. Clin. Endocrinol. Metab. 2008, 93, 3618–3626. [Google Scholar] [CrossRef]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired Mitochondrial Activity in the Insulin-Resistant Offspring of Patients with Type 2 Diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Creeden, J.F.; Kipp, Z.A.; Xu, M.; Flight, R.M.; Moseley, H.N.B.; Martinez, G.J.; Lee, W.H.; Alganem, K.; Imami, A.S.; McMullen, M.R.; et al. Hepatic Kinome Atlas: An in-Depth Identification of Kinase Pathways in Liver Fibrosis of Humans and Rodents. Hepatology 2022, 76, 1376–1388. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 15 Days | 30 Days | 45 Days | 60 Days | ||||
|---|---|---|---|---|---|---|---|---|
| Control | Insulin | Control | Insulin | Control | Insulin | Control | Insulin | |
| Weight (g) | 167 ± 5.83 | 162.8 ± 2.4 | 192.6 ± 6.6 | 205.8 ± 8.3 | 250 ± 10 | 251 ± 6 | 287 ± 15 | 291.4 ± 16 |
| Size (cm) | 15.6 ± 0.19 | 15.3 ± 0.20 | 17.4 ± 0.3 | 18.1 ± 0.4 | 19.4 ± 0.24 | 19.4 ± 0.5 | 21.2 ± 4.2 | 20.3 ± 1 |
| BMI | 0.69 ± 0.01 | 0.68 ± 0.02 | 0.64 ± 0.01 | 0.62 ± 0.02 | 0.67 ± 0.04 | 0.67 ± 0.02 | 0.63 ± 0.1 | 0.68 ±0.4 * |
| % Fat | 34.6 ± 0.21 | 34.2 ± 0.4 | 32.6 ± 0.4 | 31.7 ± 0.51 | 31.9 ± 0.6 | 31.9 ± 0.5 | 24.3 ± 6 | 32.6 ± 2 * |
| Fasting glucose (mg/dL) | 98.5 ± 5.1 | 116 ± 6.5 * | 93 ± 3.9 | 117 ± 4.1 * | 97 ± 5.1 | 131.6 ± 4.7 * | 93.6 ± 6.8 | 112 ± 5 * |
| Glucose 30′ (mg/dL) | 154.5 ± 4.3 | 164.6 ± 3.9 | 114.4 ± 5.1 | 143.5 ± 4.9 | 113 ± 4.3 | 159 ± 3.9 * | 119.5 ± 6.5 | 167.5 ± 4.5 * |
| Glucose 60′ (mg/dL) | 165.7 ± 5.5 | 177.2 ± 6.8 | 89.6 ± 4 | 148 ± 2.1 * | 133.3 ± 4.6 | 164.5 ± 4.1 * | 117.7 ± 3.3 | 141 ± 10 * |
| Glucose 90′ (mg/dL) | 179.3 ± 4.2 | 200.5 ± 5.1 * | 97 ± 5 | 126.5 ± 4.4 * | 108.3 ± 2.3 | 162.4 ± 5.2 * | 115 ± 8 | 188.5 ± 5.5 * |
| Glucose AUC | 13,775 | 15,020 | 8,970 | 12,398 * | 10,470 | 14,118 * | 10,245 | 14,065 * |
| Insulin fasting (µUI/mL) | 43.3 ± 3.4 | 51.0 ± 4.5 | 12.5 ± 1.6 | 16.5 ± 2.2 | 12.2 ± 1.2 | 14.1 ± 2.4 | 9.7 ± 1 | 12.2 ± 0.6 * |
| Insulin 30′ (µUI/mL) | 52.1 ± 4.1 | 56.3 ± 2.5 | 13.1 ± 2.5 | 15.6 ± 2.1 | 19 ± 2.2 | 20.2 ± 1.9 | 11.7 ± 1.7 | 23.8 ± 0.7 * |
| Insulin 60′ (µUI/mL) | 64.8 ± 3.8 | 67.5 ± 4.0 | 12.1 ± 2.3 | 20.1 ± 3.4* | 17.1 ± 1.5 | 28.1 ± 2.3 * | 11.5 ± 1 | 33.8 ± 1.3 * |
| Insulin 90′ (µUI/mL) | 69.7 ± 5.1 | 72.5 ± 3.4 | 9.2 ± 1.5 | 22.5 ± 1.1 * | 13.1 ± 2.3 | 18.5 ± 2.4 * | 10.3 ± 0.6 | 23.8 ± 0.9 * |
| Insulin AUC | 5205 | 5291 | 1112 | 1635 * | 1469 | 1694 * | 1000 | 1762 * |
| HOMA-IR | 1.0 ± 0.04 | 1.6 ± 0.07 | 0.45 ± 0.01 | 0.73 ± 0.04 * | 0.49 ± 0.03 | 0.68 ± 0.08 * | 0.36 ± 0.02 | 0.58 ± 0.01 * |
| QUICKY | 0.28 ± 01 | 0.26 ± 0.06 | 0.40 ± 0.03 | 0.31 ± 0.001 * | 0.38 ± 0.04 | 0.30 ± 0.01 * | 0.36 ± 0.07 | 0.32 ± 0.01 * |
| Matsuda-DeFronzo | 1.6 ± 0.06 | 1.35 ± 0.06 | 1.3 ± 0.02 | 0.90 ± 0.08 * | 6.63 ± 0.03 | 4.04 ± 0.19 * | 7.61 ± 1.1 | 4.26 ± 0.1 * |
| HIS | 4.25 ± 0.26 | 2.71 ± 0.32 * | 16.6 ± 0.84 | 10.21 ± 0.6 * | 15.3 ± 0.9 | 11.2 ± 1 * | 16.5 ± 3.1 | 10.1 ± 0.4 * |
| LIRI | 1.1 ± 0.08 | 1.3 ± 0.08 | 0.28 ± 0.13 | 0.48 ± 0.05 | 0.24 ± 0.2 | 0.39 ± 0.01* | 0.2 ± 0.02 | 0.41 ± 0.02 * |
| Parameter | 15 Days | 30 Days | 45 Days | 60 Days | ||||
|---|---|---|---|---|---|---|---|---|
| Control | Insulin | Control | Insulin | Control | Insulin | Control | Insulin | |
| Triglycerides | 77 ± 2.37 | 51.2 ± 1.59 * | 95.3 ± 2.2 | 68.2 ± 3.5 * | 100 ± 4.3 | 125 ± 6.7 * | 97 ± 4.8 | 123 ± 4.9 * |
| VLDL | 15.4 ± 0.47 | 10.2 ± 0.32 * | 19 ± 0.4 | 13.6 ± 0.71 * | 21.5 ± 0.9 | 25 ± 1.3 * | 19.4 ± 0.9 | 24 ± 1.1 * |
| ApoB | 10.5± 1.70 | 9.8± 1.16 | 9.7 ± 1 | 8.3 ± 0.8 | 11.6 ± 1 | 10.6 ± 1.5 | 10.9 ± 0.96 | 13.1 ±0.8 * |
| HDL | 42.8 ± 1.1 | 32.8 ± 2.8 * | 65 ± 2.2 | 55 ± 1.5 * | 70.2 ± 2 | 63.7 ± 2.8 * | 50.6 ± 1.2 | 43 ± 2 * |
| FFA | 7.8 ± 0.33 | 10.4 ± 0.49 * | 9.1 ± 0.08 | 11.6 ± 0.4 * | 7.1 ± 0.5 | 7.7 ± 0.2 * | 9.9 ± 0.2 | 11.8 ± 0.24 * |
| 15 Days | 30 Days | 45 Days | 60 Days | |||||
|---|---|---|---|---|---|---|---|---|
| Control | Insulin | Control | Insulin | Control | Insulin | Control | Insulin | |
| Total Glutathione (μM/mg of protein) | 19.1 ± 2.2 | 15.4 ± 2.1 | 18.9 ± 2.5 | 15.0 ± 1.4 | 18.5 ± 2.4 | 16.7 ± 1.9 | 18.9 ± 1.2 | 20.3 ± 1.4 |
| GSH (μM/mg of protein) | 13.0 ± 1.3 | 2.5 ± 0.7 * | 12.6 ± 1.8 | 2.7 ± 1.5 * | 13.3 ± 1.7 | 5.5 ± 1.4 * | 12.9 ± 1.6 | 11 ± 1.3 |
| GSSG (μM/mg of protein) | 6.2 ± 0.8 | 14.8 ± 1.0 * | 6.3 ± 0.6 | 12.7 ± 1.5 * | 6.8 ± 0.9 | 11.6 ± 1.1 * | 6.5 ± 1.1 | 10.3 ± 1.7 * |
| 2GSH/GSSG | 4.2 ± 1.0 | 0.3 ± 0.09 * | 4.0 ± 0.5 | 0.4 ± 0.02 * | 4.1 ± 0.4 | 0.6 ± 0.9 * | 4.2 ± 0.8 | 2.2 ± 0.7 * |
| GPx (U min−1/mg of protein) | 2.6 ± 0.1 | 3.1 ± 0.1 | 2.3 ± 0.1 | 2.9 ± 0.2 | 3.1 ± 0.4 | 3.2 ± 0.8 | 2.9 ± 0.2 | 3.3 ± 0.4 |
| GR (U min−1/mg of protein) | 1016.2 ± 77 | 1555.9 ± 160 * | 1147.7 ± 170 | 1759.6 ± 218 * | 1051 ± 134 | 1654 ± 121 * | 1152 ± 181 | 1893 ± 162 * |
| GT (U min−1/mg of protein) | 839.9 ± 81 | 988.1 ± 123 | 658.2 ± 92 | 958.7 ± 126 | 758.2 ± 140 | 896.4 ± 132 | 792 ± 164 | 784.4 ± 170 |
| SOD (U min−1/mg of protein) | 8.0 ± 0.4 | 7.1 ± 0.5 | 7.6 ± 0.9 | 7.2 ± 0.5 | 7.7 ± 0.3 | 7.5 ± 0.4 | 7.9 ± 0.9 | 7.3 ± 1.4 |
| CAT (U min−1/mg of protein) | 19.9 ± 2.4 | 18.0 ± 2.3 | 19.5 ± 1.8 | 18.4 ± 2.4 | 19.1 ± 1.9 | 18.5 ± 1.3 | 20.1 ± 1.4 | 19.7 ± 2.3 |
| MT (μg/mg of protein) | 1.84 ± 0.2 | 2.4 ± 0.3* | 1.3 ± 0.3 | 2.5 ± 0.5 * | 1.9 ± 0.3 | 2.7 ± 0.7* | 1.7 ± 0.4 | 2.6 ± 0.2 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarmiento-Ortega, V.E.; Moroni-González, D.; Diaz, A.; García-González, M.Á.; Brambila, E.; Treviño, S. Hepatic Insulin Resistance Model in the Male Wistar Rat Using Exogenous Insulin Glargine Administration. Metabolites 2023, 13, 572. https://doi.org/10.3390/metabo13040572
Sarmiento-Ortega VE, Moroni-González D, Diaz A, García-González MÁ, Brambila E, Treviño S. Hepatic Insulin Resistance Model in the Male Wistar Rat Using Exogenous Insulin Glargine Administration. Metabolites. 2023; 13(4):572. https://doi.org/10.3390/metabo13040572
Chicago/Turabian StyleSarmiento-Ortega, Victor Enrique, Diana Moroni-González, Alfonso Diaz, Miguel Ángel García-González, Eduardo Brambila, and Samuel Treviño. 2023. "Hepatic Insulin Resistance Model in the Male Wistar Rat Using Exogenous Insulin Glargine Administration" Metabolites 13, no. 4: 572. https://doi.org/10.3390/metabo13040572
APA StyleSarmiento-Ortega, V. E., Moroni-González, D., Diaz, A., García-González, M. Á., Brambila, E., & Treviño, S. (2023). Hepatic Insulin Resistance Model in the Male Wistar Rat Using Exogenous Insulin Glargine Administration. Metabolites, 13(4), 572. https://doi.org/10.3390/metabo13040572