
Epigenetic Modifications Induced by the Gut Microbiota May Result from What We Eat: Should We Talk about Precision Diet in Health and Disease?

Abstract
1. Introduction
2. Gut Microbiota
2.1. The Impact of Diet on Gut Microbiota
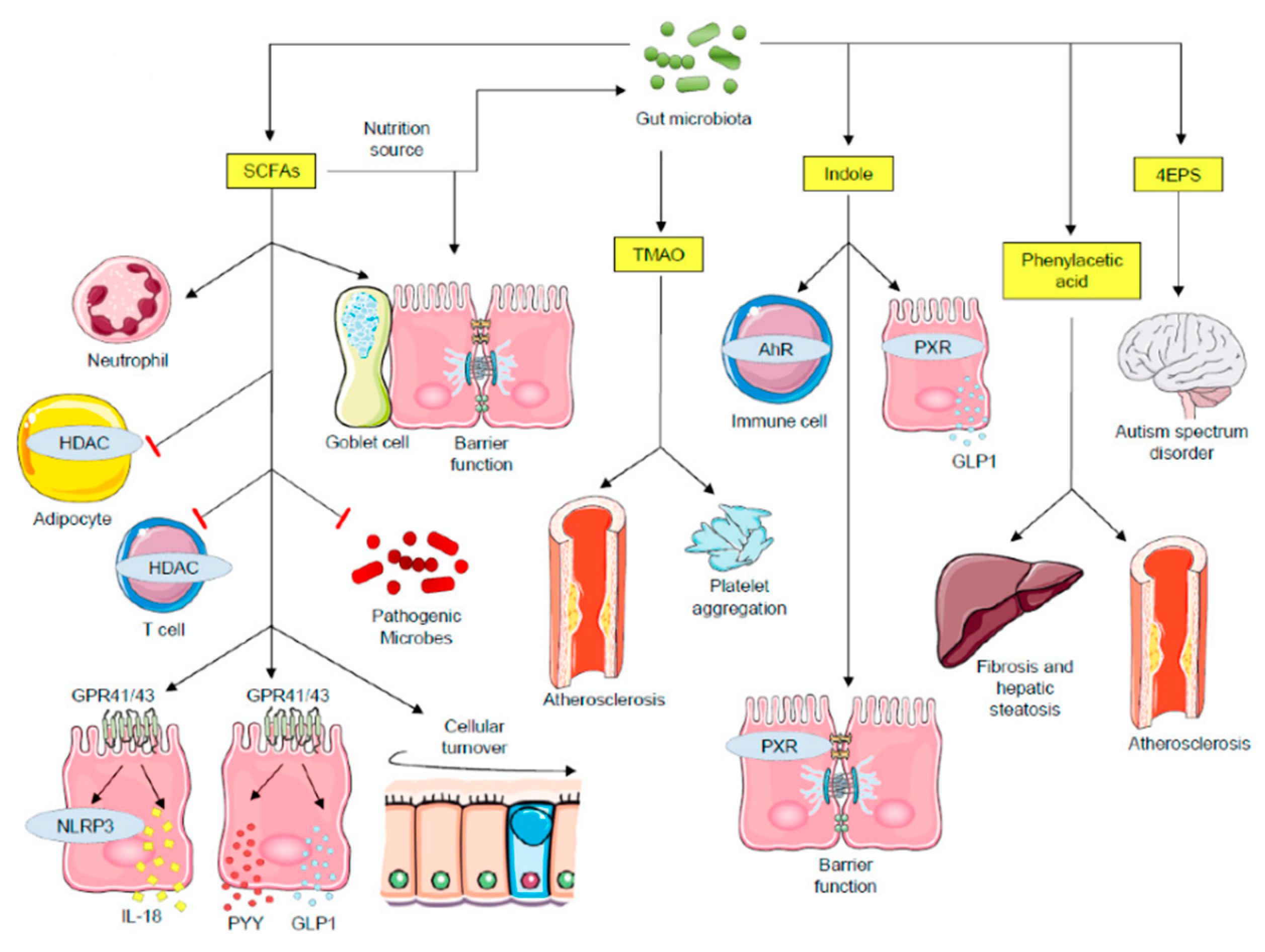
2.2. Microbial Metabolites as Critical Transducers of Microbiota–Host Signaling
2.3. Epigenetics and Epigenetic Modifications in Disease
2.4. Microbiota Metabolites Modulate the Activity of the Enzymes Involved in Dynamic Chromatin Modifications
3. The Therapeutic Potential of the Diet–Microbiota–Epigenetics Triade
4. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Allum, F.; Grundberg, E. Capturing functional epigenomes for insight into metabolic diseases. Mol. Metab. 2020, 38, 100936. [Google Scholar] [CrossRef] [PubMed]
- Alam, R.; Abdolmaleky, H.M.; Zhou, J.-R. Microbiome, inflammation, epigenetic alterations, and mental diseases. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2017, 174, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Sun, L.; Zhang, X. Integration of microbiome and epigenome to decipher the pathogenesis of autoimmune diseases. J. Autoimmun. 2017, 83, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Chang, H.-W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Zheng, D.; Elinav, E. Diet–microbiota interactions and personalized nutrition. Nat. Rev. Microbiol. 2019, 17, 742–753. [Google Scholar] [CrossRef]
- Feinberg, A.P. Epigenetics at the Epicenter of Modern Medicine. JAMA 2008, 299, 1345–1350. [Google Scholar] [CrossRef]
- Hullar, M.A.J.; Fu, B.C. Diet, the Gut Microbiome, and Epigenetics. Cancer J. 2014, 20, 170–175. [Google Scholar] [CrossRef]
- Tiffon, C. The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease. Int. J. Mol. Sci. 2018, 19, 3425. [Google Scholar] [CrossRef]
- Wang, Z.; Long, H.; Chang, C.; Zhao, M.; Lu, Q. Crosstalk between metabolism and epigenetic modifications in autoimmune diseases: A comprehensive overview. Cell. Mol. Life Sci. 2018, 75, 3353–3369. [Google Scholar] [CrossRef]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef] [PubMed]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Bultman, S.J. Interplay between diet, gut microbiota, epigenetic events, and colorectal cancer. Mol. Nutr. Food Res. 2017, 61, 1500902. [Google Scholar] [CrossRef]
- Krautkramer, K.A.; Kreznar, J.H.; Romano, K.A.; Vivas, E.I.; Barrett-Wilt, G.A.; Rabaglia, M.E.; Keller, M.P.; Attie, A.D.; Rey, F.E.; Denu, J.M. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol. Cell 2016, 64, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Food, Immunity, and the Microbiome. Gastroenterology 2015, 148, 1107–1119. [Google Scholar] [CrossRef]
- Gago, B.; Nyström, T.; Cavaleiro, C.; Rocha, B.S.; Barbosa, R.M.; Laranjinha, J.; Lundberg, J.O. The potent vasodilator ethyl nitrite is formed upon reaction of nitrite and ethanol under gastric conditions. Free. Radic. Biol. Med. 2008, 45, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Schnorr, S.L.; Candela, M.; Rampelli, S.; Centanni, M.; Consolandi, C.; Basaglia, G.; Turroni, S.; Biagi, E.; Peano, C.; Severgnini, M.; et al. Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 2014, 5, 3654. [Google Scholar] [CrossRef]
- Santacruz, A.; Marcos, A.; Wärnberg, J.; Martí, A.; Martin-Matillas, M.; Campoy, C.; Moreno, L.A.; Veiga, O.; Redondo-Figuero, C.; Garagorri, J.M.; et al. Interplay Between Weight Loss and Gut Microbiota Composition in Overweight Adolescents. Obesity 2009, 17, 1906–1915. [Google Scholar] [CrossRef]
- Zimmer, J.; Lange, B.J.; Frick, J.-S.; Sauer, H.; Zimmermann, K.; Schwiertz, A.; A Rusch, K.; Klosterhalfen, S.; Enck, P. A vegan or vegetarian diet substantially alters the human colonic faecal microbiota. Eur. J. Clin. Nutr. 2011, 66, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, E.D.; Smits, S.A.; Tikhonov, M.; Higginbottom, S.K.; Wingreen, N.S.; Sonnenburg, J.L. Diet-induced extinctions in the gut microbiota compound over generations. Nature 2016, 529, 212–215. [Google Scholar] [CrossRef] [PubMed]
- D’Aquila, P.; Carelli, L.L.; De Rango, F.; Passarino, G.; Bellizzi, D. Gut Microbiota as Important Mediator Between Diet and DNA Methylation and Histone Modifications in the Host. Nutrients 2020, 12, 597. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.C.; Ramos-Lopez, O.; Riezu-Boj, J.I.; Milagro, F.I.; Martinez, J.A. Diet, Gut Microbiota, and Obesity: Links with Host Genetics and Epigenetics and Potential Applications. Adv. Nutr. 2019, 10, S17–S30. [Google Scholar] [CrossRef]
- Kaczmarek, J.L.; Thompson, S.V.; Holscher, H. Complex interactions of circadian rhythms, eating behaviors, and the gastrointestinal microbiota and their potential impact on health. Nutr. Rev. 2017, 75, 673–682. [Google Scholar] [CrossRef]
- Bhat, M.I.; Kapila, R. Dietary metabolites derived from gut microbiota: Critical modulators of epigenetic changes in mammals. Nutr. Rev. 2017, 75, 374–389. [Google Scholar] [CrossRef]
- Rauf, A.; Khalil, A.A.; Rahman, U.-U.; Khalid, A.; Naz, S.; Shariati, M.A.; Rebezov, M.; Urtecho, E.Z.; de Albuquerque, R.D.D.G.; Anwar, S.; et al. Recent advances in the therapeutic application of short-chain fatty acids (SCFAs): An updated review. Crit. Rev. Food Sci. Nutr. 2021, 62, 6034–6054. [Google Scholar] [CrossRef]
- Ríos-Covián, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; De Los Reyes-Gavilán, C.G.; Salazar, N. Intestinal Short Chain Fatty Acids and their Link with Diet and Human Health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef]
- Hiippala, K.; Jouhten, H.; Ronkainen, A.; Hartikainen, A.; Kainulainen, V.; Jalanka, J.; Satokari, R. The Potential of Gut Commensals in Reinforcing Intestinal Barrier Function and Alleviating Inflammation. Nutrients 2018, 10, 988. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; A Wade, P. Crosstalk between the microbiome and epigenome: Messages from bugs. J. Biochem. 2017, 163, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, Y. Gut microbiota derived metabolites in cardiovascular health and disease. Protein Cell 2018, 9, 416–431. [Google Scholar] [CrossRef]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef]
- Roberts, A.B.; Gu, X.; Buffa, J.A.; Hurd, A.G.; Wang, Z.; Zhu, W.; Gupta, N.; Skye, S.M.; Cody, D.B.; Levison, B.S.; et al. Development of a gut microbe–targeted nonlethal therapeutic to inhibit thrombosis potential. Nat. Med. 2018, 24, 1407–1417. [Google Scholar] [CrossRef]
- Hoyles, L.; Fernández-Real, J.-M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H.; et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080. [Google Scholar] [CrossRef]
- Kawabata, K.; Yoshioka, Y.; Terao, J. Role of Intestinal Microbiota in the Bioavailability and Physiological Functions of Dietary Polyphenols. Molecules 2019, 24, 370. [Google Scholar] [CrossRef]
- Rocha, B.S.; Laranjinha, J. Nitrate from diet might fuel gut microbiota metabolism: Minding the gap between redox signaling and inter-kingdom communication. Free. Radic. Biol. Med. 2020, 149, 37–43. [Google Scholar] [CrossRef]
- Stols-Gonçalves, D.; Tristão, L.S.; Henneman, P.; Nieuwdorp, M. Epigenetic Markers and Microbiota/Metabolite-Induced Epigenetic Modifications in the Pathogenesis of Obesity, Metabolic Syndrome, Type 2 Diabetes, and Non-alcoholic Fatty Liver Disease. Curr. Diabetes Rep. 2019, 19, 31. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.; Barnes, S.; Demark-Wahnefried, W.; Morrow, C.; Salvador, C.; Skibola, C.; Tollefsbol, T.O. Influences of diet and the gut microbiome on epigenetic modulation in cancer and other diseases. Clin. Epigenetics 2015, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fouquier, J.; Huizar, N.M.; Donnelly, J.; Glickman, C.; Kang, D.-W.; Maldonado, J.; Jones, R.A.; Johnson, K.; Adams, J.B.; Krajmalnik-Brown, R.; et al. The Gut Microbiome in Autism: Study-Site Effects and Longitudinal Analysis of Behavior Change. Msystems 2021, 6, e00848-20. [Google Scholar] [CrossRef] [PubMed]
- Onal, E.M.; Afsar, B.; Covic, A.; Vaziri, N.D.; Kanbay, M. Gut microbiota and inflammation in chronic kidney disease and their roles in the development of cardiovascular disease. Hypertens. Res. 2018, 42, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Miro-Blanch, J.; Yanes, O. Epigenetic Regulation at the Interplay Between Gut Microbiota and Host Metabolism. Front. Genet. 2019, 10, 638. [Google Scholar] [CrossRef] [PubMed]
- Gerhauser, C. Impact of dietary gut microbial metabolites on the epigenome. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170359. [Google Scholar] [CrossRef]
- Furrow, R.E.; Christiansen, F.B.; Feldman, M.W. Environment-Sensitive Epigenetics and the Heritability of Complex Diseases. Genetics 2011, 189, 1377–1387. [Google Scholar] [CrossRef]
- Chan, J.C.; Maze, I. Nothing Is Yet Set in (Hi)stone: Novel Post-Translational Modifications Regulating Chromatin Function. Trends Biochem. Sci. 2020, 45, 829–844. [Google Scholar] [CrossRef]
- Park, S.; Kim, G.W.; Kwon, S.H.; Lee, J. Broad domains of histone H3 lysine 4 trimethylation in transcriptional regulation and disease. FEBS J. 2020, 287, 2891–2902. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Z.; Yung, S.; Lu, Q. Epigenetic dynamics in immunity and autoimmunity. Int. J. Biochem. Cell Biol. 2015, 67, 65–74. [Google Scholar] [CrossRef]
- Abdul, Q.A.; Yu, B.P.; Chung, H.Y.; Jung, H.A.; Choi, J.S. Epigenetic modifications of gene expression by lifestyle and environment. Arch. Pharmacal Res. 2017, 40, 1219–1237. [Google Scholar] [CrossRef]
- Klein, B.J.; Krajewski, K.; Restrepo, S.; Lewis, P.W.; Strahl, B.D.; Kutateladze, T. Recognition of cancer mutations in histone H3K36 by epigenetic writers and readers. Epigenetics 2018, 13, 683–692. [Google Scholar] [CrossRef]
- Feinberg, A.P. Phenotypic plasticity and the epigenetics of human disease. Nature 2007, 447, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Toor, S.M.; Nair, V.S.; Elkord, E. Role of Epigenetic Modifications in Inhibitory Immune Checkpoints in Cancer Development and Progression. Front. Immunol. 2020, 11, 1469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, J.; Liu, X.; Xiao, Z.; Lei, G.; Lee, H.; Koppula, P.; Cheng, W.; Mao, C.; Zhuang, L.; et al. H2A Monoubiquitination Links Glucose Availability to Epigenetic Regulation of the Endoplasmic Reticulum Stress Response and Cancer Cell Death. Cancer Res 2020, 80, 2243–2256. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Bilokapic, S.; Halic, M. Nucleosome and ubiquitin position Set2 to methylate H3K36. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Tian, J.; Yuan, L. Sirtuin 6 inhibits colon cancer progression by modulating PTEN/AKT signaling. Biomed. Pharmacother. 2018, 106, 109–116. [Google Scholar] [CrossRef]
- Enck, P.; Aziz, Q.; Barbara, G.; Farmer, A.D.; Fukudo, S.; Mayer, E.A.; Niesler, B.; Quigley, E.M.M.; Rajilić-Stojanović, M.; Schemann, M.; et al. Irritable bowel syndrome. Nat. Rev. Dis. Prim. 2016, 2, 16014. [Google Scholar] [CrossRef]
- Kalla, R.; Ventham, N.T.; Kennedy, N.; Quintana, J.F.; Nimmo, E.R.; Buck, A.; Satsangi, J. MicroRNAs: New players in IBD. Gut 2014, 64, 504–513. [Google Scholar] [CrossRef]
- Shahgaldi, S.; Kahmini, F.R. A comprehensive review of Sirtuins: With a major focus on redox homeostasis and metabolism. Life Sci. 2021, 282, 119803. [Google Scholar] [CrossRef] [PubMed]
- Etchegaray, J.-P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Akondy, R.S.; Fitch, M.; Edupuganti, S.; Yang, S.; Kissick, H.T.; Li, K.W.; Youngblood, B.A.; Abdelsamed, H.A.; McGuire, D.J.; Cohen, K.W.; et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature 2017, 552, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Dimri, M.; Bommi, P.; Sahasrabuddhe, A.; Khandekar, J.D.; Dimri, G.P. Dietary omega-3 polyunsaturated fatty acids suppress expression of EZH2 in breast cancer cells. Carcinog. 2009, 31, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, A.E.; Baniasadi, M.; Giansiracusa, D.; Giansiracusa, M.; Garcia, M.; Fryda, Z.; Wong, T.L.; Bishayee, A. Sulforaphane: A Broccoli Bioactive Phytocompound with Cancer Preventive Potential. Cancers 2021, 13, 4796. [Google Scholar] [CrossRef]
- Miyamoto, H.; Tatsukawa, T.; Shimohata, A.; Yamagata, T.; Suzuki, T.; Amano, K.; Mazaki, E.; Raveau, M.; Ogiwara, I.; Oba-Asaka, A.; et al. Impaired cortico-striatal excitatory transmission triggers epilepsy. Nat. Commun. 2019, 10, 1917. [Google Scholar] [CrossRef]
- Hung, S.; Kuo, K.; Wu, C.; Tarng, D. Indoxyl Sulfate: A Novel Cardiovascular Risk Factor in Chronic Kidney Disease. J. Am. Heart Assoc. 2017, 6, e005022. [Google Scholar] [CrossRef]
- Gentile, C.L.; Weir, T.L. The gut microbiota at the intersection of diet and human health. Science 2018, 362, 776–780. [Google Scholar] [CrossRef]
- De Angelis, M.; Garruti, G.; Minervini, F.; Bonfrate, L.; Portincasac, P.; Gobbetti, M. The Food-gut Human Axis: The Effects of Diet on Gut Microbiota and Metabolome. Curr. Med. Chem. 2019, 26, 3567–3583. [Google Scholar] [CrossRef]
- De Filippis, F.; Pellegrini, N.; Vannini, L.; Jeffery, I.B.; La Storia, A.; Laghi, L.; Serrazanetti, D.I.; Di Cagno, R.; Ferrocino, I.; Lazzi, C.; et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut Microbiota 2016, 65, 1812–1821. [Google Scholar] [CrossRef]
- García-Montero, C.; Fraile-Martínez, O.; Gómez-Lahoz, A.; Pekarek, L.; Castellanos, A.; Noguerales-Fraguas, F.; Coca, S.; Guijarro, L.; García-Honduvilla, N.; Asúnsolo, A.; et al. Nutritional Components in Western Diet Versus Mediterranean Diet at the Gut Microbiota–Immune System Interplay. Implications for Health and Disease. Nutrients 2021, 13, 699. [Google Scholar] [CrossRef] [PubMed]
- Thakur, C.; Chen, F. Connections between metabolism and epigenetics in cancers. Semin. Cancer Biol. 2019, 57, 52–58. [Google Scholar] [CrossRef]
- Laranjinha, J.; Cadenas, E. Redox Cycles of Caffeic Acid, alpha Tocopherol, and Ascorbate: Implications for Protection of Lo—Density Lipoproteins Against Oxidation. IUBMB Life 1999, 48, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Rocha, B.S.; Nunes, C.; Pereira, C.; Barbosa, R.M.; Laranjinha, J. A shortcut to wide-ranging biological actions of dietary polyphenols: Modulation of the nitrate–nitrite–nitric oxide pathway in the gut. Food Funct. 2014, 5, 1646–1652. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Görlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef]
- González-Sarrías, A.; Larrosa, M.; Tomás-Barberán, F.A.; Dolara, P.; Espín, J.C. NF-kappaB-dependent anti-inflammatory activity of urolithins, gut microbiota ellagic acid-derived metabolites, in human colonic fibroblasts. Br. J. Nutr. 2010, 104, 503–512. [Google Scholar] [CrossRef]
- El-Wetidy, M.S.; Ahmad, R.; Rady, I.; Helal, H.; Rady, M.I.; Vaali-Mohammed, M.-A.; Al-Khayal, K.; Traiki, T.B.; Abdulla, M.-H. Urolithin A induces cell cycle arrest and apoptosis by inhibiting Bcl-2, increasing p53-p21 proteins and reactive oxygen species production in colorectal cancer cells. Cell Stress Chaperones 2021, 26, 473–493. [Google Scholar] [CrossRef]
- Hutchinson, A.; Tingö, L.; Brummer, R. The Potential Effects of Probiotics and ω-3 Fatty Acids on Chronic Low-Grade Inflammation. Nutrients 2020, 12, 2402. [Google Scholar] [CrossRef]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-κB. J. Am. Heart Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef]
- Chen, M.-L.; Yi, L.; Zhang, Y.; Zhou, X.; Ran, L.; Yang, J.; Zhu, J.-D.; Zhang, Q.-Y.; Mi, M.-T. Resveratrol Attenuates Trimethylamine- N -Oxide (TMAO)-Induced Atherosclerosis by Regulating TMAO Synthesis and Bile Acid Metabolism via Remodeling of the Gut Microbiota. Mbio 2016, 7, e02210-15. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Nutritional Behavior | Microbiotic Structure | References |
|---|---|---|
| Omnivorous | ↑ Bifidobacterium and Bacteroides species, Escherichia coli, and Enterobacteriaceae. | [6,15] |
| Vegans/ vegetarian | ↑ Coliforms (vegan). ↑ Prevotella (vegetarian). ↓ Bacteroides, Bifidobacteria, Escherichia coli and Enterobacteriaceae. | [23,25] |
| Diet high in animal protein (temporary) | ↑ Bacteroides, Alistipes, Bilophila and Clostridia. ↓ Firmutes (Eubacterium rectale, Ruminococcus bromii and Roseburia species) and Bifidobacterium. | [1,5,6,26] |
| Diet high in plant protein (temporary) | ↑ Bifidobacteria and commensal Lactobacilli; ↓ Bacteroides and Clostridium perfringens. | [5,6] |
| Diet high in resistant starch (temporary) | ↑ Proportions of Firmicutes bacteria related to Ruminococcus bromii. | [15] |
| Diet rich in unsaturated fat | ↑ Lactobacillus, Streptococcus, Bifidobacteria and Akkermansia muciniphila. | [5] |
| Diet rich in saturated fat | ↑ Bacteroides, Bilophila, Faecalibacterium prausnitzii. | [5] |
| Diet high in fiber | ↑ Bacterial abundance; ↑ Bacteroides, Bifidobacterium, Lactobacilli, Roseburia, Eubacteria and Ruminococcus ↓ Enterococcus and Clostridium species | [5,6,26] |
| Diet rich in polyphenols | ↑ Bifidobacteria and Lactobacilli; ↓ Bacteroides, Clostridia, Salmonella typhimurium and Staphylococcus aureus. | [5] |
| Rural diet | ↑ Bacteroidetes (including the genera Xylanibacter and Prevotella); ↑ microbiotic variety. | [1,6,8] |
| Urbanized diet | Loss of Treponema species; loss of microbiota diversity. | [1,6] |
| Temporary calorie- restrictive diet | ↓ Blautia coccoides. ↑ Bacteroides. | [1] |
| Gut Microbiota Metabolite | Metabolite Producing Bacteria | Dietary Component of Origin | References |
|---|---|---|---|
| Short-chain fatty acids (SCFAs) | Faecalibacterium prausnitzii, Bifidobacterium spp., Lactobacillus spp., Roseburia spp., Eubacterium hallii. | Dietary fiber (undigested complex carbohydrates) | [4,5,6,8,14,31] |
| Trimethylamine N- oxide (TMAO) | Proteus mirabillis. Bacteria present in higher abundances in omnivores populations. | L-carnitine, choline and phosphatidylcholine (particularly from red meat) | [1,32,33,34,35] |
| Indole and indole derivates | Escherichia coli, Clostridium spp. and Bacteroides spp. Enterococcus faecalis. | Tryptophan | [6,34,36,37] |
| 4-hydroxyphenyl- acetic acid and 4–ethylphenyl- sulfate (4EPS) | Species largely unknown. | Tyrosine | [6,34] |
| Phenylacetic acid | Species largely unknown. | Phenylalanine | [6,34,38] |
| Isothiocyanate | Escherichia coli, Bifidobacterium sp., Bacteroides thetaiotaomicron, Enterococcus faecium, Enterococcus faecalis and Peptostreptococcus sp. | Glucosinolates | [8] |
| Methylated catechins, phenolic products, and ring fission products (ex. valerolactone) | Species largely unknown. | Catechins | [8,39] |
| Urolithin | C. coccoides, Bifidobacterium spp., and Lactobacillus spp. | Ellagic acid | [8,13,39] |
| Dihydrogenistein, dihydrodaidzein, equol, enterolactone, enterodiol and O-desmethylangolensin (O-DMA) | Lactococcus garvieae, Eggerthella sp. YY7918, Adlercreutzia equolifaciens, Slackia isoflavoniconvertens, Slackia equolifaciens, Slackia sp. NATTS. | Phytoestrogens | [13,34] |
| Protocatechuic acid (PCA) | Species largely unknown. | Anthocyanins | [34,39] |
| Nitrite | Species largely unknown, although studies point out Actinobacteria and Firmicutes as highest nitrate-reducers. | Nitrate | [40] |
| Disease | Epigenetic Modifications | References |
|---|---|---|
| Colorectal cancer | Epigenetic modifications targeting several genes, including APC, GATA4, MLH1 and p16INK4a. | [13] |
| Irritable bowel syndrome | Diminished miR-199 level, correlating with TRPV1 (transient receptor potential cation channel subfamily V member 1) upregulation and increased visceral sensitivity. | [59] |
| Inflammatory bowel disease | MiRNA dysregulation in Th17 cells, affecting its function and differentiation. | [60] |
| Obesity | Several obesity-related genes with differential methylation, including CD36, CLDN1, HAND2, HOXC6, SORBS2 and PPARG; H3K9me in white adipose tissue regarding differentiation from white to brown adipose cells. | [41] |
| Non-alcoholic fatty liver disease (NAFLD) | PNPLA3 (patatin-like phospholipase domain containing 3) gene hypermethylation. | [41] |
| Lupus erythematosus | Overexpression of various genes in CD4+ T cells, like ITGAL, PRF1, TNFSF7, TNFSF5, leading to the activation of B cells and over production of autoantibodies. | [50] |
| Rheumatoid arthritis | Patients with RA have lowered DNA methyltransferase expression in Treg cells and significantly reduced DNA methylation in the Foxp3 promoter. There are other interesting findings in the existing studies [50]. | [50] |
| Type 1 diabetes mellitus (T1DM) | Several aberrant DNA and histone modifications. Regarding DNA methylation, examples include increased methylation in the insulin-like growth factor-binding protein 1 (IGFBP1) gene, leading to increased levels of circulating IGFBP1; hypermethylation of promoter regions of Interleukin-2 receptor alfa chain gene; hypermethylation of Foxp3 gene promoter; etc. An example of histone aberrant epigenetic modifications are HDACs reduced expression in CD4+ T cells. | [50] |
| Type 2 diabetes mellitus (T2DM) | H3K27me3 modification in myocytes, downregulating genes responsible for muscle function and upregulating genes involved in T2D inflammation. Several genes related to risk of T2D with aberrant DNA methylation, including FTO, KCNQ1, IRS1, TCFL2 and THADA. | [41] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reva, K.; Laranjinha, J.; Rocha, B.S. Epigenetic Modifications Induced by the Gut Microbiota May Result from What We Eat: Should We Talk about Precision Diet in Health and Disease? Metabolites 2023, 13, 375. https://doi.org/10.3390/metabo13030375
Reva K, Laranjinha J, Rocha BS. Epigenetic Modifications Induced by the Gut Microbiota May Result from What We Eat: Should We Talk about Precision Diet in Health and Disease? Metabolites. 2023; 13(3):375. https://doi.org/10.3390/metabo13030375
Chicago/Turabian StyleReva, Katerina, João Laranjinha, and Bárbara S. Rocha. 2023. "Epigenetic Modifications Induced by the Gut Microbiota May Result from What We Eat: Should We Talk about Precision Diet in Health and Disease?" Metabolites 13, no. 3: 375. https://doi.org/10.3390/metabo13030375
APA StyleReva, K., Laranjinha, J., & Rocha, B. S. (2023). Epigenetic Modifications Induced by the Gut Microbiota May Result from What We Eat: Should We Talk about Precision Diet in Health and Disease? Metabolites, 13(3), 375. https://doi.org/10.3390/metabo13030375