
Development of an Untargeted Metabolomics Strategy to Study the Metabolic Rewiring of Dendritic Cells upon Lipopolysaccharide Activation

 , , and
, , and
Abstract
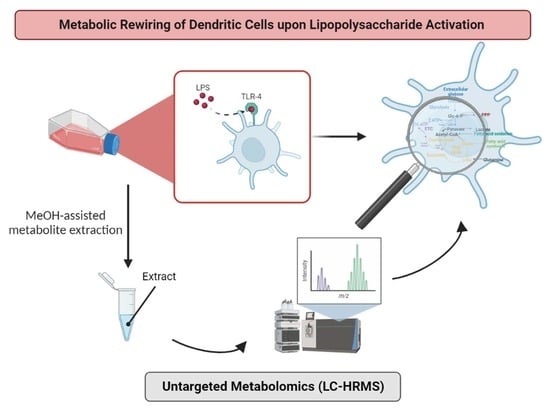
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
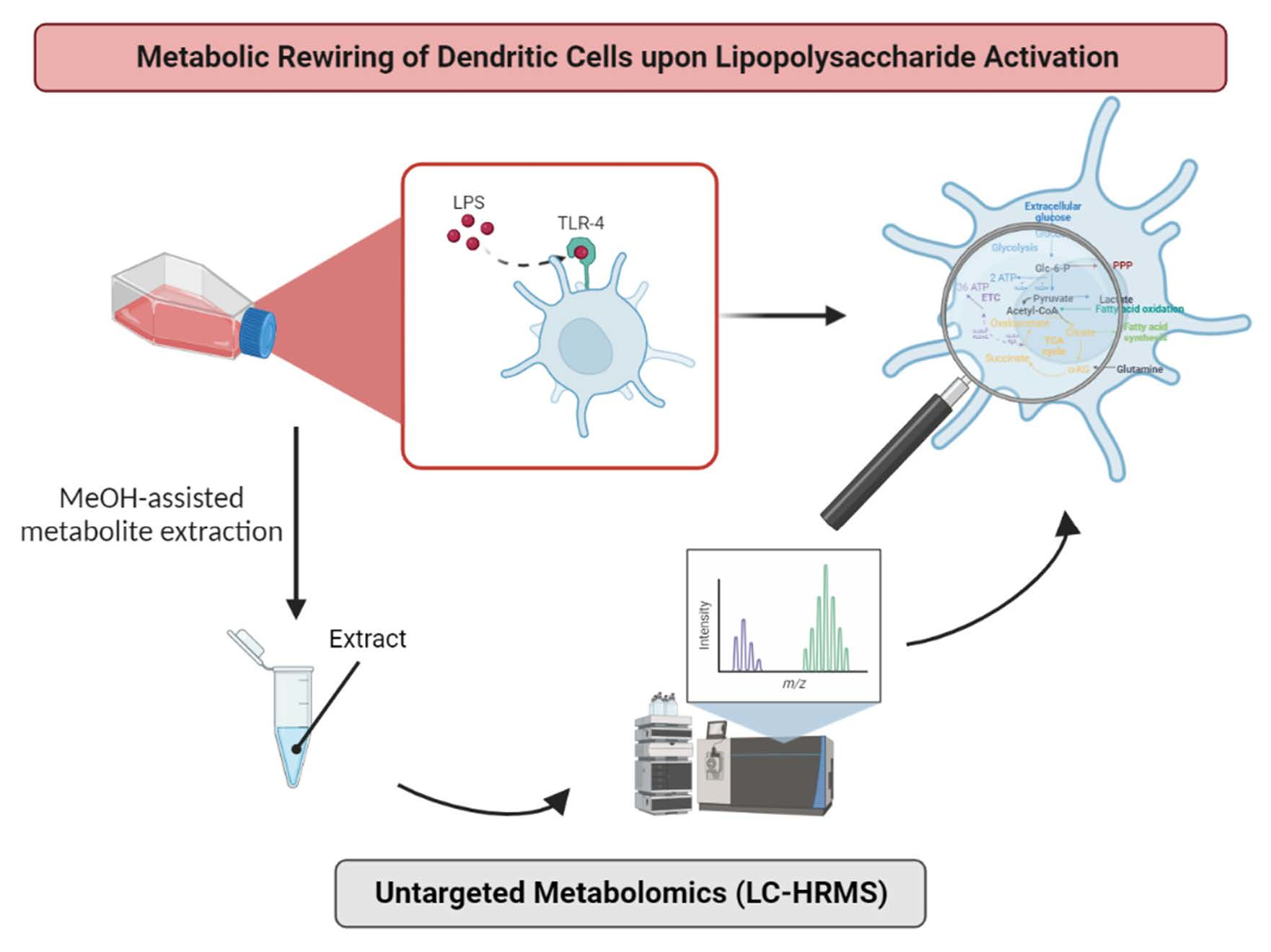
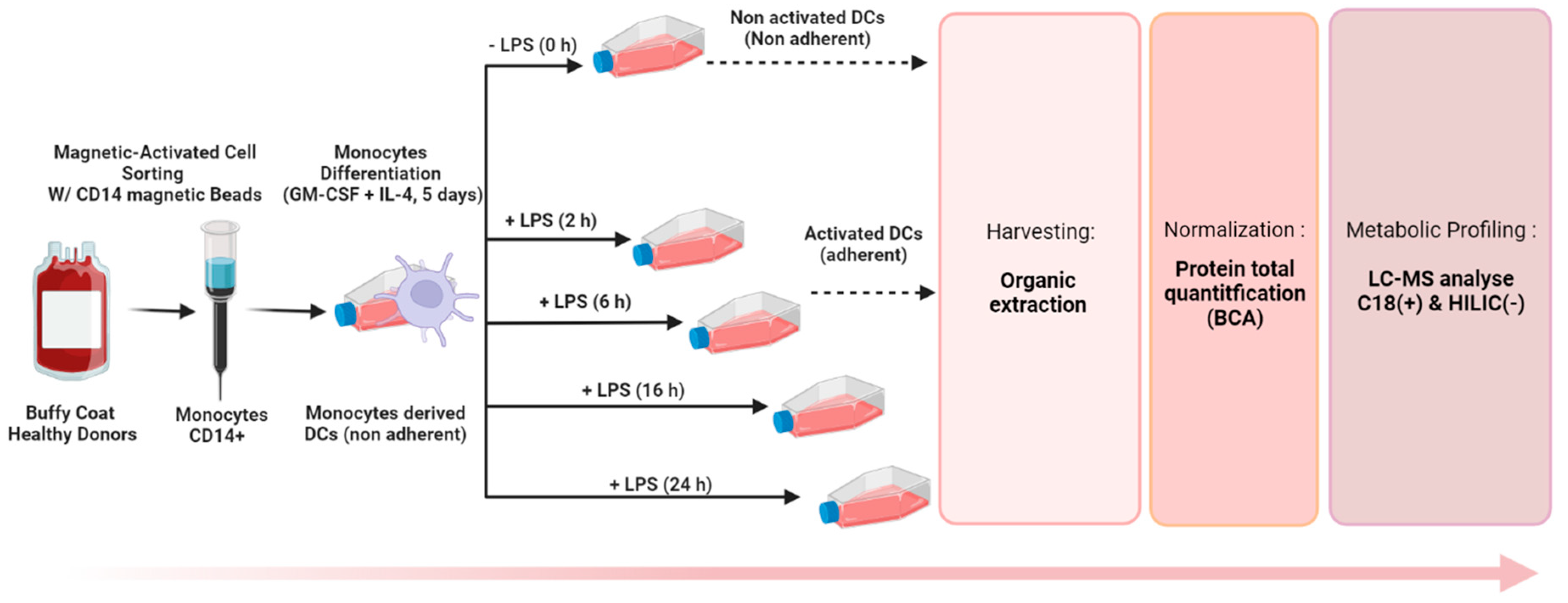
2. Experimental Design
2.1. Chemicals
2.2. Generation of Monocyte-Derived Dendritic Cells
2.3. Flow Cytometry Phenotyping of DCs
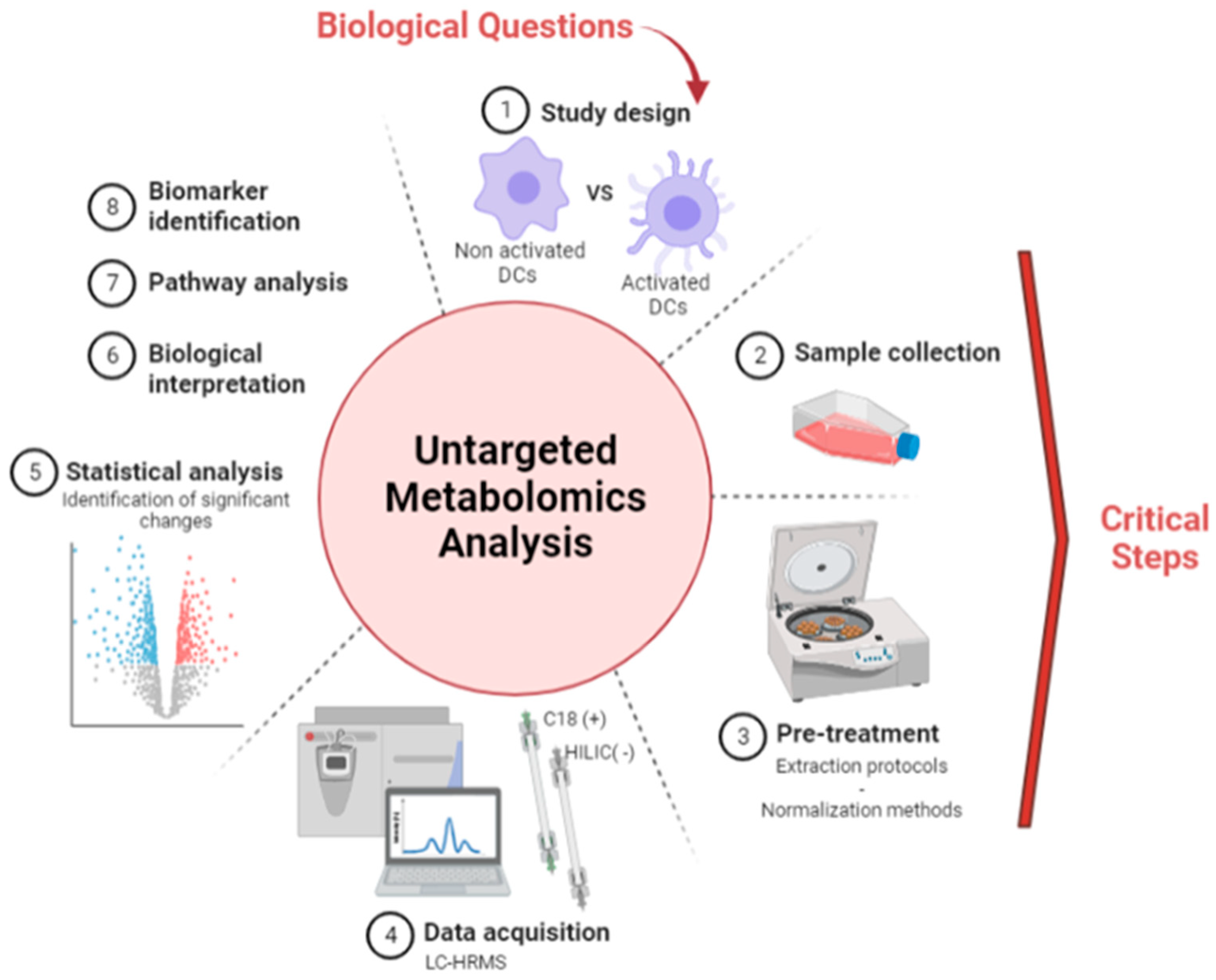
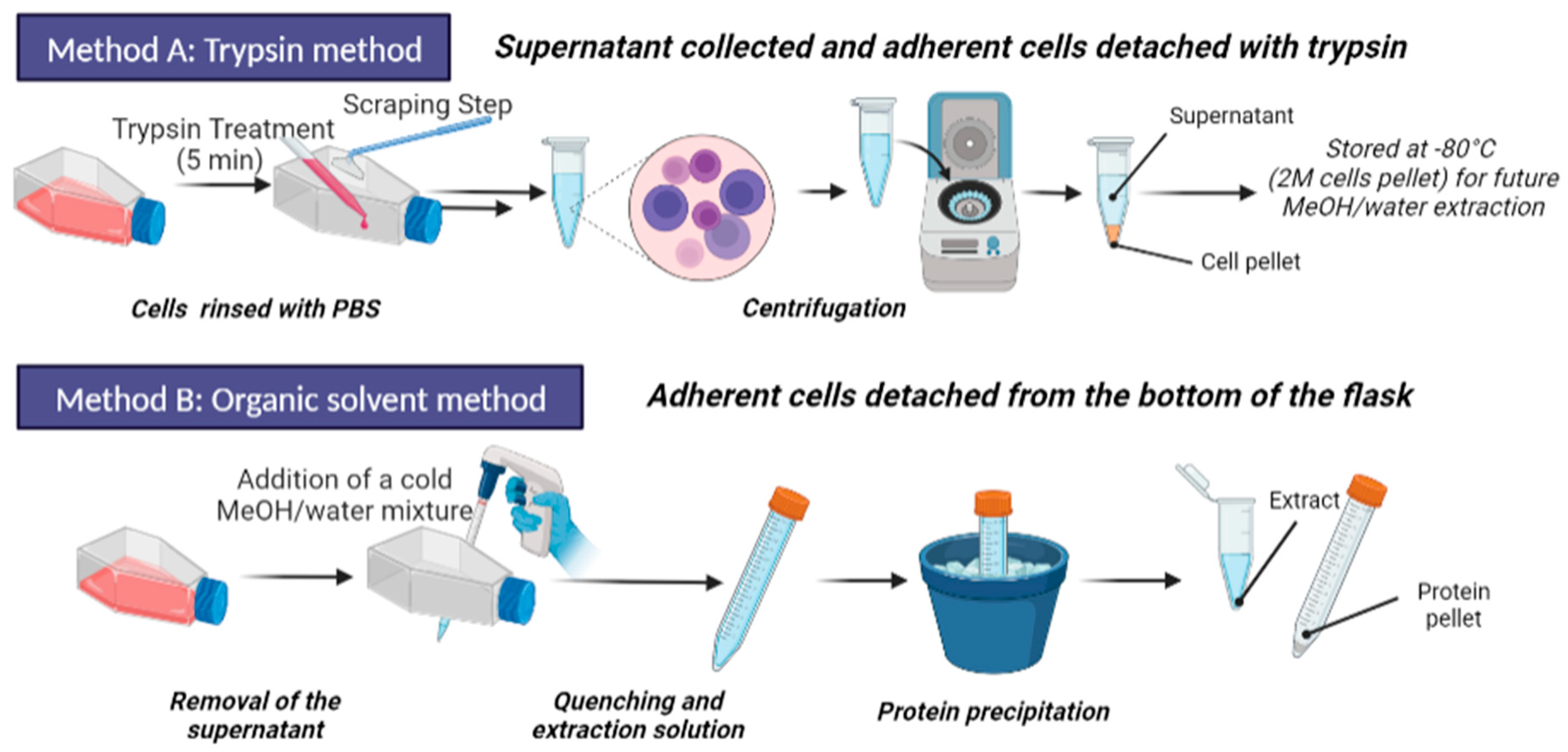
2.4. Sample Preparation Step
2.4.1. Trypsination and Metabolite Extraction
2.4.2. Organic Solvent Extraction and Quenching
2.5. Total Protein Quantification
2.6. LC-HRMS Analyses
2.7. Statistical Analyses
3. Results and Discussion
3.1. Optimization of Sample Preparation and Normalization Protocols
3.1.1. Development of a Sample Normalization Procedure
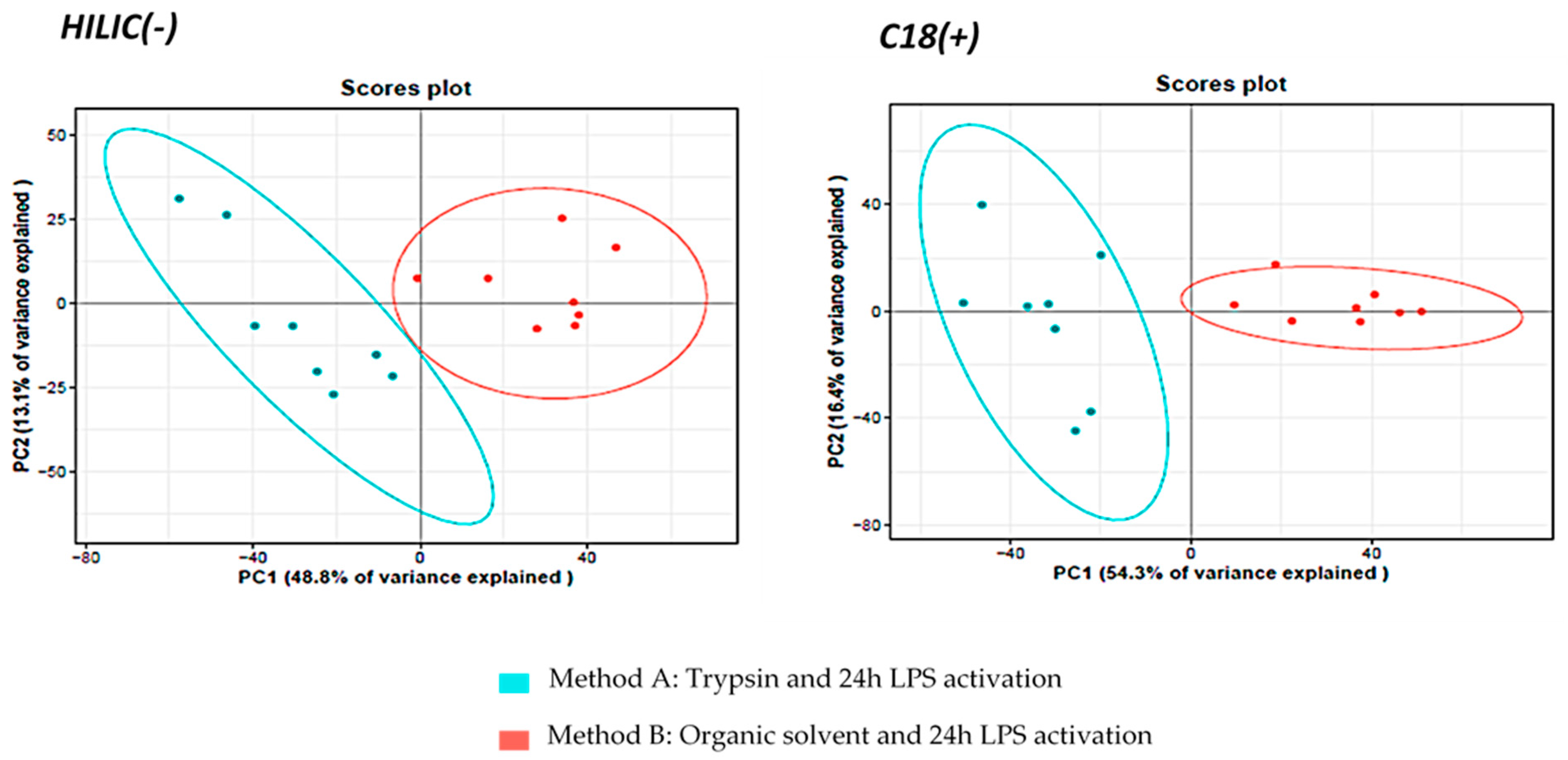
3.1.2. Comparison of Metabolite Fingerprints Obtained with Trypsin- or Organic Solvents-Based Protocols
3.2. Metabolomics Investigation of the Reprogramming Process of DCs upon LPS Exposure
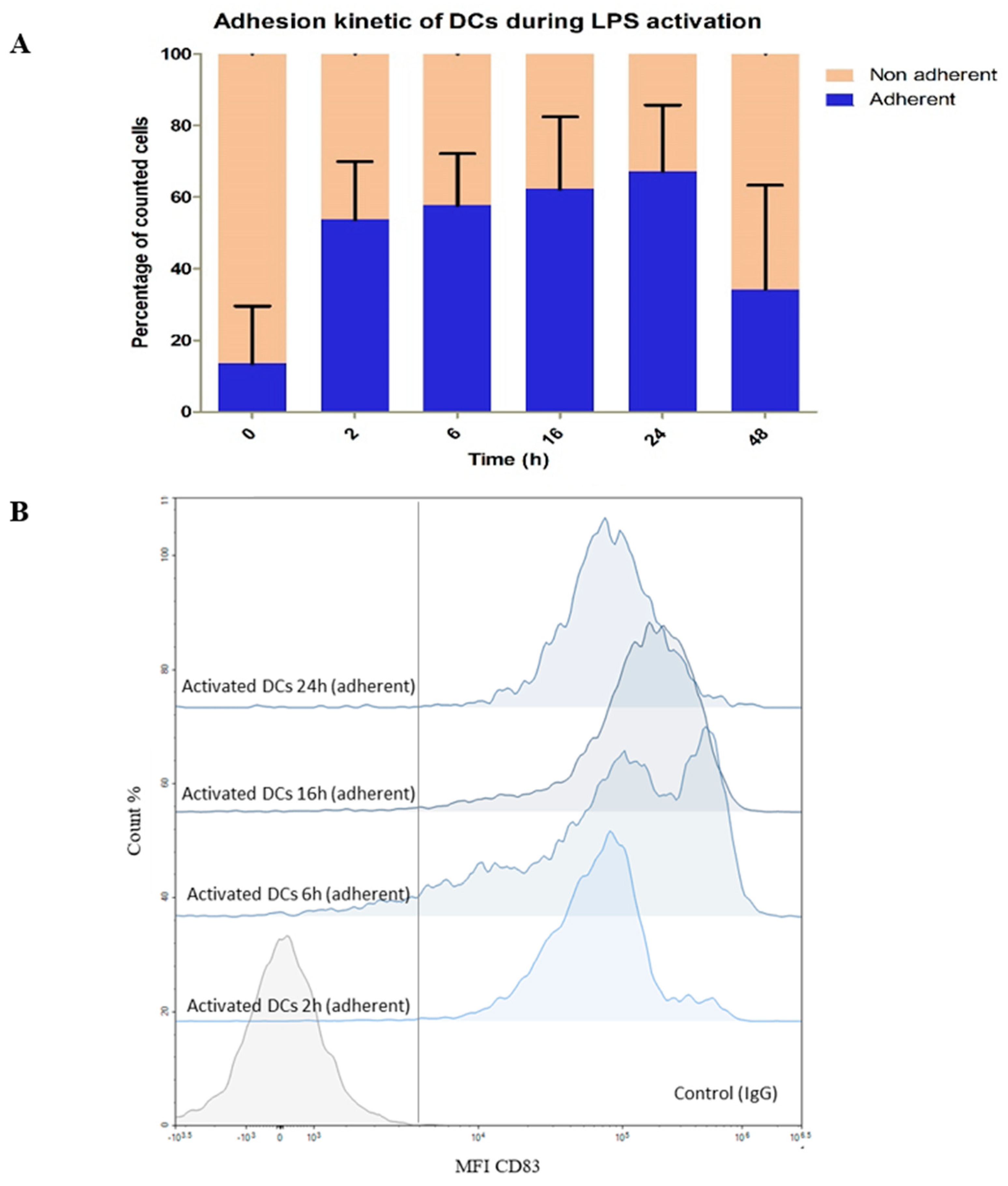
3.2.1. Impact of LPS on DC Phenotype
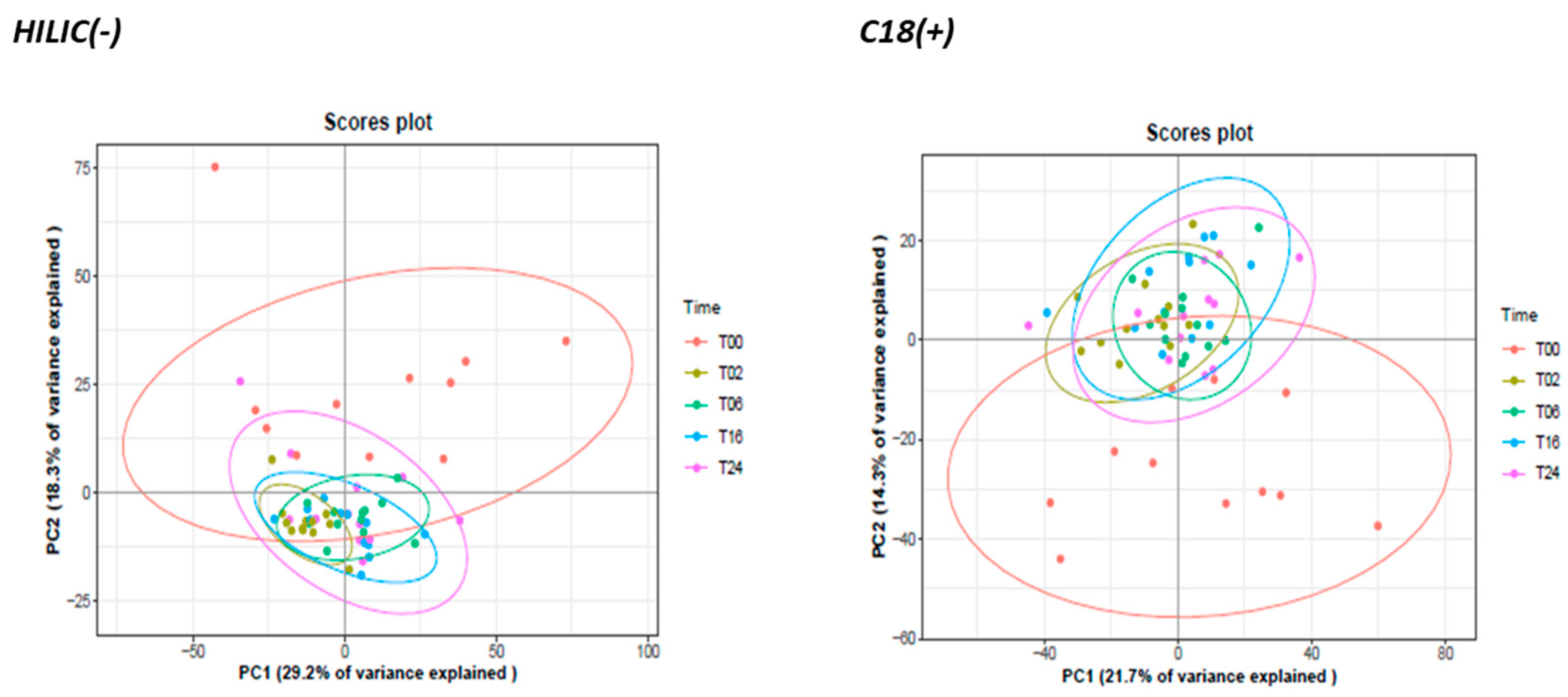
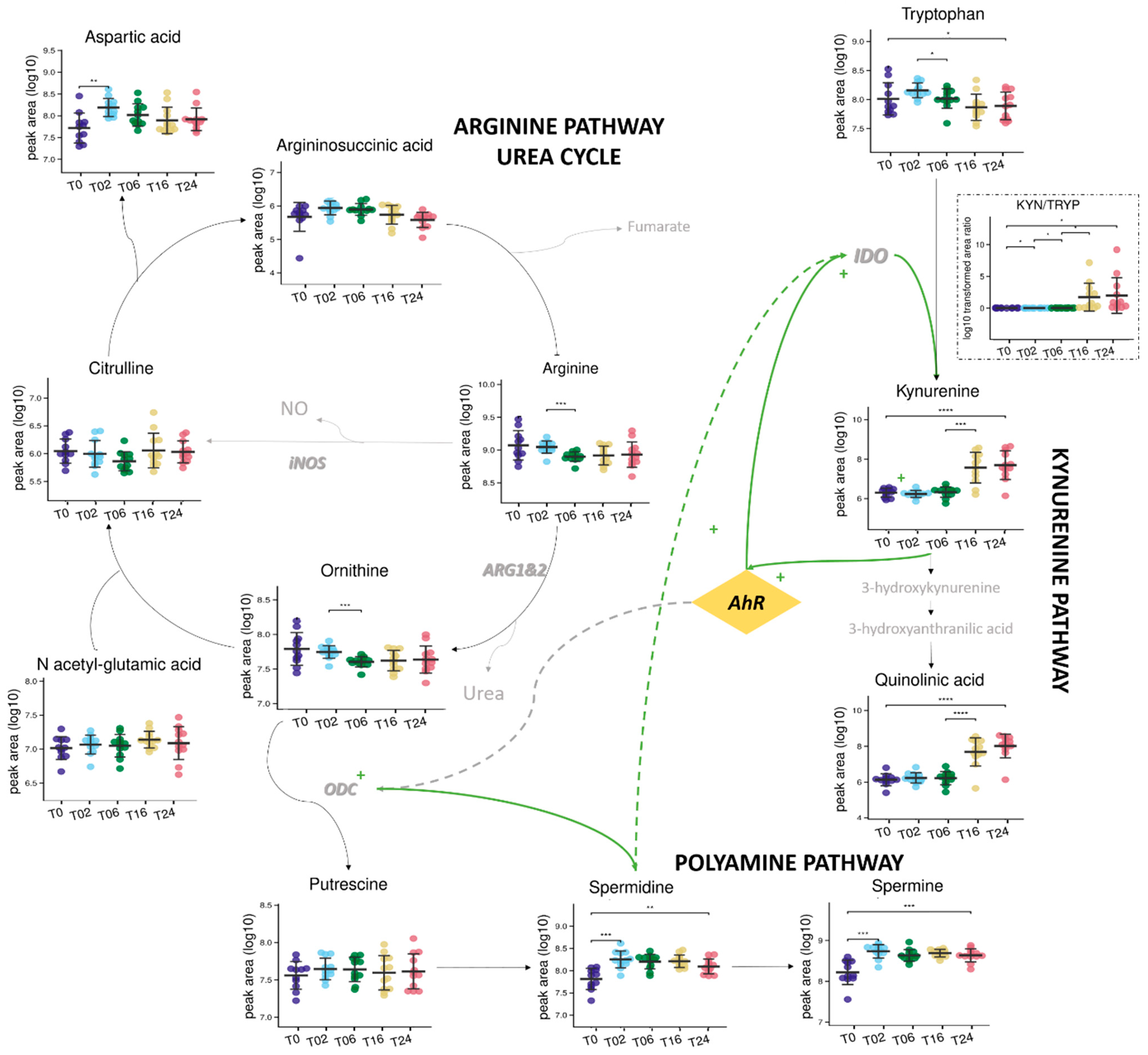
3.2.2. Monitoring of DCs and Metabolic Reprogramming upon LPS Stimulation by Untargeted Metabolomics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roca, M.; Alcoriza, M.I.; Garcia-Cañaveras, J.C.; Lahoz, A. Reviewing the metabolome coverage provided by LC-MS: Focus on sample preparation and chromatography-A tutorial. Anal. Chim. Acta 2021, 1147, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Sands, C.J.; Gómez-Romero, M.; Correia, G.; Chekmeneva, E.; Camuzeaux, S.; Izzi-Engbeaya, C.; Dhillo, W.S.; Takats, Z.; Lewis, M.R. Representing the Metabolome with High Fidelity: Range and Response as Quality Control Factors in LC-MS-Based Global Profiling. Anal. Chem. 2021, 93, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Krausz, K.W.; Manna, S.K.; Li, F.; Johnson, C.H.; Gonzalez, F.J. Optimization of harvesting, extraction, and analytical protocols for UPLC-ESI-MS-based metabolomic analysis of adherent mammalian cancer cells. Anal. Bioanal. Chem. 2013, 405, 5279–5289. [Google Scholar] [CrossRef]
- Muschet, C.; Möller, G.; Prehn, C.; de Angelis, M.H.; Adamski, J.; Tokarz, J. Removing the bottlenecks of cell culture metabolomics: Fast normalization procedure, correlation of metabolites to cell number, and impact of the cell harvesting method. Metabolomics 2016, 12, 151. [Google Scholar] [CrossRef] [PubMed]
- Domenick, T.M.; Gill, E.L.; Vedam-Mai, V.; Yost, R.A. Mass Spectrometry-Based Cellular Metabolomics: Current Approaches, Applications, and Future Directions. Anal. Chem. 2021, 93, 546–566. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, K.; Nürnberger, N.; Kaspar, H.; Gruber, M.A.; Almstetter, M.F.; Oefner, P.J. Metabolite extraction from adherently growing mammalian cells for metabolomics studies: Optimization of harvesting and extraction protocols. Anal. Bioanal. Chem. 2011, 399, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Granucci, F.; Ferrero, E.; Foti, M.; Aggujaro, D.; Vettoretto, K.; Ricciardi-Castagnoli, P. Early events in dendritic cell maturation induced by LPS. Microbes Infect. 1999, 1, 1079–1084. [Google Scholar] [CrossRef]
- Yi, H.-J.; Lu, G.-X. Adherent and non-adherent dendritic cells are equivalently qualified in GM-CSF, IL-4 and TNF-a culture system. Cell. Immunol. 2012, 277, 44–48. [Google Scholar] [CrossRef]
- Appel, S. The Culture Dish Surface Influences the Phenotype and Cytokine Production of Human Monocyte-Derived Dendritic Cells. Front. Immunol. 2019, 10, 10. [Google Scholar]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C. Innate immune recognition: Mechanisms and pathways: Innate immune recognition. Immunol. Rev. 2000, 173, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.B.; Reinert, L.S.; Paludan, S.R. Innate recognition of intracellular pathogens: Detection and activation of the first line of defense. APMIS 2009, 117, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Rattigan, K.M.; Pountain, A.; Regnault, C.; Achcar, F.; Vincent, I.; Goodyear, C.S.; Barrett, M.P. Metabolomic profiling of macrophages determines the discrete metabolomic signature and metabolomic interactome triggered by polarising immune stimuli. PLoS ONE 2018, 13, e0194126. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Loftus, R.M.; Finlay, D.K. Immunometabolism: Cellular Metabolism Turns Immune Regulator. J. Biol. Chem. 2016, 291, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Rathmell, J.; Pearce, E. SnapShot: Immunometabolism. Cell Metab. 2015, 22, 190–190.e1. [Google Scholar] [CrossRef]
- Haas, R.; Smith, J.; Rocher-Ros, V.; Nadkarni, S.; Montero-Melendez, T.; D’Acquisto, F.; Bland, E.J.; Bombardieri, M.; Pitzalis, C.; Perretti, M.; et al. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol. 2015, 13, e1002202. [Google Scholar] [CrossRef]
- Harber, K.J.; De Goede, K.E.; Verberk, S.G.S.; Meinster, E.; De Vries, H.E.; Van Weeghel, M.; De Winther, M.P.J.; Bossche, J.V.D. Succinate Is an Inflammation-Induced Immunoregulatory Metabolite in Macrophages. Metabolites 2020, 10, 372. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Williams, N.C.; O’Neill, L.A.J. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 2018, 9, 141. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, D.; Zheng, X.-L.; Tang, C. Itaconate: An emerging determinant of inflammation in activated macrophages. Immunol. Cell Biol. 2018, 97, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.R.; Namboodiri, M.A. Tryptophan and the immune response. Immunol. Cell Biol. 2003, 81, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.R.; Arun, P.; Puthillathu, N.; Vengilote, R.; Ives, J.A.; Badawy, A.A.; Namboodiri, A.M. Quinolinate as a Marker for Kynurenine Metabolite Formation and the Unresolved Question of NAD+ Synthesis During Inflammation and Infection. Front. Immunol. 2020, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Mok, H.J.; Lee, D.Y.; Park, S.C.; Kim, G.S.; Lee, S.E.; Lee, Y.S.; Kim, K.P.; Kim, H.D. UPLC-QqQ/MS-Based Lipidomics Approach To Characterize Lipid Alterations in Inflammatory Macrophages. J. Proteome Res. 2017, 16, 1460–1469. [Google Scholar] [CrossRef]
- Chanput, W.; Mes, J.J.; Savelkoul, H.F.J.; Wichers, H.J. Characterization of polarized THP-1 macrophages and polarizing ability of LPS and food compounds. Food Funct. 2013, 4, 266–276. [Google Scholar] [CrossRef]
- Fall, F.; Lamy, E.; Brollo, M.; Naline, E.; Lenuzza, N.; Thévenot, E.; DeVillier, P.; Grassin-Delyle, S. Metabolic reprograming of LPS-stimulated human lung macrophages involves tryptophan metabolism and the aspartate-arginosuccinate shunt. PLoS ONE 2020, 15, e0230813. [Google Scholar] [CrossRef]
- Sindelar, M.; Patti, G.J. Chemical Discovery in the Era of Metabolomics. J. Am. Chem. Soc. 2020, 142, 9097–9105. [Google Scholar] [CrossRef]
- Roux, A.; Lison, D.; Junot, C.; Heilier, J.-F. Applications of liquid chromatography coupled to mass spectrometry-based metabolomics in clinical chemistry and toxicology: A review. Clin. Biochem. 2011, 44, 119–135. [Google Scholar] [CrossRef]
- Giacomoni, F.; Le Corguillé, G.; Monsoor, M.; Landi, M.; Pericard, P.; Pétéra, M.; Duperier, C.; Tremblay-Franco, M.; Martin, J.-F.; Jacob, D.; et al. Workflow4Metabolomics: A collaborative research infrastructure for computational metabolomics. Bioinformatics 2015, 31, 1493–1495. [Google Scholar] [CrossRef]
- Boudah, S.; Olivier, M.-F.; Aros-Calt, S.; Oliveira, L.; Fenaille, F.; Tabet, J.-C.; Junot, C. Annotation of the human serum metabolome by coupling three liquid chromatography methods to high-resolution mass spectrometry. J. Chromatogr. B 2014, 966, 34–47. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Barbier Saint Hilaire, P.; Rousseau, K.; Seyer, A.; Dechaumet, S.; Damont, A.; Junot, C.; Fenaille, F. Comparative Evaluation of Data Dependent and Data Independent Acquisition Workflows Implemented on an Orbitrap Fusion for Untargeted Metabolomics. Metabolites 2020, 10, 158. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O.; Robertson, D.; Griffin, J.; van der Werf, M.; Nikolau, B.; Morrison, N.; Sumner, L.W.; Goodacre, R.; Hardy, N.W.; Taylor, C.; et al. The metabolomics standards initiative (MSI). Metabolomics 2007, 3, 175–178. [Google Scholar] [CrossRef]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 1994, 179, 1109–1118. [Google Scholar] [CrossRef]
- Luo, X.; Gu, X.; Li, L. Development of a simple and efficient method of harvesting and lysing adherent mammalian cells for chemical isotope labeling LC-MS-based cellular metabolomics. Anal. Chim. Acta 2018, 1037, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhang, X.; Bai, Y.; Yang, L.; Wu, T.; Zhang, J.; Li, L.; Zhang, H.; Wang, H. Optimization of the sample preparation method for adherent cell metabolomics based on ultra-performance liquid chromatography coupled to mass spectrometry. Anal. Methods 2019, 11, 3678–3686. [Google Scholar] [CrossRef]
- Luo, Y.; Geng, N.; Zhang, B.; Chen, J.; Zhang, H. Effects of harvesting and extraction methods on metabolite recovery from adherently growing mammalian cells. Anal. Methods 2020, 12, 2491–2498. [Google Scholar] [CrossRef]
- Chen, S.; So, E.C.; Strome, S.E.; Zhang, X. Impact of Detachment Methods on M2 Macrophage Phenotype and Function. J. Immunol. Methods 2015, 426, 56–61. [Google Scholar] [CrossRef]
- Willmann, L.; Schlimpert, M.; Halbach, S.; Erbes, T.; Stickeler, E.; Kammerer, B. Metabolic profiling of breast cancer: Differences in central metabolism between subtypes of breast cancer cell lines. J. Chromatogr. B 2015, 1000, 95–104. [Google Scholar] [CrossRef]
- Abdul-Hamid, N.A.; Abas, F.; Maulidiani, M.; Ismail, I.S.; Tham, C.L.; Swarup, S.; Umashankar, S. NMR metabolomics for evaluating passage number and harvesting effects on mammalian cell metabolome. Anal. Biochem. 2019, 576, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Teng, Q.; Huang, W.; Collette, T.W.; Ekman, D.R.; Tan, C. A direct cell quenching method for cell-culture based metabolomics. Metabolomics 2009, 5, 199–208. [Google Scholar] [CrossRef]
- Silva, L.P.; Lorenzi, P.L.; Purwaha, P.; Yong, V.; Hawke, D.H.; Weinstein, J.N. Measurement of DNA Concentration as a Normalization Strategy for Metabolomic Data from Adherent Cell Lines. Anal. Chem. 2013, 85, 9536–9542. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-Y.; Liao, H.-W.; Tsai, I.-L.; Tseng, Y.J.; Kuo, C.-H. Using the Matrix-Induced Ion Suppression Method for Concentration Normalization in Cellular Metabolomics Studies. Anal. Chem. 2015, 87, 9731–9739. [Google Scholar] [CrossRef]
- Lovrien, R.; Matulis, D. Assays for Total Protein. Curr. Protoc. Protein Sci. 1995, 1, 3.4.1–3.4.24. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.; Fiehn, O.; Goodacre, R.; Lane, A.N.; et al. Proposed minimum reporting standards for chemical analysis: Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- Comte, B.; Monnerie, S.; Brandolini-Bunlon, M.; Canlet, C.; Castelli, F.; Chu-Van, E.; Colsch, B.; Fenaille, F.; Joly, C.; Jourdan, F.; et al. Multiplatform metabolomics for an integrative exploration of metabolic syndrome in older men. EBioMedicine 2021, 69, 103440. [Google Scholar] [CrossRef]
- Imbert, A.; Rompais, M.; Selloum, M.; Castelli, F.; Mouton-Barbosa, E.; Brandolini-Bunlon, M.; Chu-Van, E.; Joly, C.; Hirschler, A.; Roger, P.; et al. ProMetIS, deep phenotyping of mouse models by combined proteomics and metabolomics analysis. Sci. Data 2021, 8, 311. [Google Scholar] [CrossRef]
- Moreau, R.; Clària, J.; Aguilar, F.; Fenaille, F.; Lozano, J.J.; Junot, C.; Colsch, B.; Caraceni, P.; Trebicka, J.; Pavesi, M.; et al. Blood metabolomics uncovers inflammation-associated mitochondrial dysfunction as a potential mechanism underlying ACLF. J. Hepatol. 2020, 72, 688–701. [Google Scholar] [CrossRef]
- Rushing, B.R.; Schroder, M.; Sumner, S.C.J. Comparison of Lysis and Detachment Sample Preparation Methods for Cultured Triple-Negative Breast Cancer Cells Using UHPLC–HRMS-Based Metabolomics. Metabolites 2022, 12, 168. [Google Scholar] [CrossRef]
- Sallusto, F.; Cella, M.; Danieli, C.; Lanzavecchia, A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: Downregulation by cytokines and bacterial products. J. Exp. Med. 1995, 182, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Savina, A.; Amigorena, S. Phagocytosis and antigen presentation in dendritic cells. Immunol. Rev. 2007, 219, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Abdi, K.; Singh, N.J.; Matzinger, P. Lipopolysaccharide-Activated Dendritic Cells: “Exhausted” or Alert and Waiting? J. Immunol. 2012, 188, 5981–5989. [Google Scholar] [CrossRef]
- Sugimoto, M.; Sakagami, H.; Yokote, Y.; Onuma, H.; Kaneko, M.; Mori, M.; Sakaguchi, Y.; Soga, T.; Tomita, M. Non-targeted metabolite profiling in activated macrophage secretion. Metabolomics 2012, 8, 624–633. [Google Scholar] [CrossRef]
- Abuawad, A.; Mbadugha, C.; Ghaemmaghami, A.M.; Kim, D.-H. Metabolic characterisation of THP-1 macrophage polarisation using LC–MS-based metabolite profiling. Metabolomics 2020, 16, 33. [Google Scholar] [CrossRef]
- de Macedo, L.H.; Souza CO, S.; Gardinassi, L.G.; Faccioli, L.H. CD14 regulates the metabolomic profiles of distinct macrophage subsets under steady and activated states. Immunobiology 2022, 227, 152191. [Google Scholar] [CrossRef]
- Mondanelli, G.; Bianchi, R.; Pallotta, M.T.; Orabona, C.; Albini, E.; Iacono, A.; Belladonna, M.L.; Vacca, C.; Fallarino, F.; Macchiarulo, A.; et al. A Relay Pathway between Arginine and Tryptophan Metabolism Confers Immunosuppressive Properties on Dendritic Cells. Immunity 2017, 46, 233–244. [Google Scholar] [CrossRef]
- Soulet, D.; Rivest, S. Polyamines play a critical role in the control of the innate immune response in the mouse central nervous system. J. Cell Biol. 2003, 162, 257–268. [Google Scholar] [CrossRef]
- Li, G.; Ding, H.; Yu, X.; Meng, Y.; Li, J.; Guo, Q.; Zhou, H.; Shen, N. Spermidine Suppresses Inflammatory DC Function by Activating the FOXO3 Pathway and Counteracts Autoimmunity. Iscience 2020, 23, 100807. [Google Scholar] [CrossRef]
- Proietti, E.; Rossini, S.; Grohmann, U.; Mondanelli, G. Polyamines and Kynurenines at the Intersection of Immune Modulation. Trends Immunol. 2020, 41, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Pallotta, M.T.; Rossini, S.; Suvieri, C.; Coletti, A.; Orabona, C.; Macchiarulo, A.; Volpi, C.; Grohmann, U. Indoleamine 2,3-dioxygenase 1 (IDO1): An up-to-date overview of an eclectic immunoregulatory enzyme. FEBS J. 2022, 289, 6099–6118. [Google Scholar] [CrossRef] [PubMed]
- Sorgdrager, F.J.H.; Naudé, P.J.W.; Kema, I.P.; Nollen, E.A.; De Deyn, P.P. Tryptophan Metabolism in Inflammaging: From Biomarker to Therapeutic Target. Front. Immunol. 2019, 10, 2565. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Hanieh, H.; Nakahama, T.; Kishimoto, T. The roles of aryl hydrocarbon receptor in immune responses. Int. Immunol. 2013, 25, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michieletto, J.; Delvaux, A.; Chu-Van, E.; Junot, C.; Fenaille, F.; Castelli, F.A. Development of an Untargeted Metabolomics Strategy to Study the Metabolic Rewiring of Dendritic Cells upon Lipopolysaccharide Activation. Metabolites 2023, 13, 311. https://doi.org/10.3390/metabo13030311
Michieletto J, Delvaux A, Chu-Van E, Junot C, Fenaille F, Castelli FA. Development of an Untargeted Metabolomics Strategy to Study the Metabolic Rewiring of Dendritic Cells upon Lipopolysaccharide Activation. Metabolites. 2023; 13(3):311. https://doi.org/10.3390/metabo13030311
Chicago/Turabian StyleMichieletto, Jessica, Aurélie Delvaux, Emeline Chu-Van, Christophe Junot, François Fenaille, and Florence A. Castelli. 2023. "Development of an Untargeted Metabolomics Strategy to Study the Metabolic Rewiring of Dendritic Cells upon Lipopolysaccharide Activation" Metabolites 13, no. 3: 311. https://doi.org/10.3390/metabo13030311
APA StyleMichieletto, J., Delvaux, A., Chu-Van, E., Junot, C., Fenaille, F., & Castelli, F. A. (2023). Development of an Untargeted Metabolomics Strategy to Study the Metabolic Rewiring of Dendritic Cells upon Lipopolysaccharide Activation. Metabolites, 13(3), 311. https://doi.org/10.3390/metabo13030311