4. Remorse
It might not have escaped the reader’s attention that we do not seriously propose using Mad Hatter for any practical compound annotation. Instead, Mad Hatter was solely developed for the purpose of demonstrating common issues in computational method development and evaluation. No real-world data should ever be analyzed with Mad Hatter. Besides choosing outrageous features for Mad Hatter, we deliberately introduced a series of blunders in our evaluations that allowed us to reach the reported annotation rates. We start by describing the blunders, and we discuss the outrageous features in the next section. Unfortunately, the mentioned blunders can regularly be found in other scientific publications that present novel in silico methods; since this is not blame-and-shame, we will refrain from explicitly pointing out those papers. We argue that it is rather editors and reviewers than authors who are responsible for the current unpleasant situation. The blunders we deliberately made in the development and evaluation of Mad Hatter may serve as a guide to editors and reviewers for spotting similar issues “in the wild”.
Early publications [
16,
18,
19] went through a great deal of effort to demonstrate and evaluate not only the power but also the limitations of the presented methods. In contrast, it lately became a habit to demonstrate that a new method strongly out-competes all others in some evaluations. It is sad to say that those claims are often pulverized later in blind CASMI challenges. One way to achieve the desired outcome is to carefully select our competitors; leave out those that are hard to beat. Another possibility is to evaluate other methods ourselves but not invest too much time into this part of the evaluation; nobody will mind the inexplicable performance drop of the other method unless we explicitly mention it. A third possibility is to restrict reported results to one evaluation dataset where the desired outcome is reached. Finally, we may simply present results as if our method was the first to ever deal with the presented problem, and simply ignore the presence of other methods; this is the approach we chose for
Mad Hatter. Yet, the most common trick to make our method shine is to evaluate using the data our method already knows from training. This is commonly referred to as “data leakage”.
Do not evaluate using your training data. For the Mad Hatter evaluation, the first blunder to notice is that we do not differentiate between the training set and the dataset used for evaluation. Consequently, the machine learning model is rewarded for memorizing instead of learning. This must be regarded as a show-stopper for a method publication, as all numbers reported are basically useless. In fact, CASMI 2016 offers two independent datasets (train and test) that can be used for exactly this purpose. If we repeat the above evaluation but this time, make a clear distinction between training and test set, correct annotations drop to 61.4% using the large neural network. This might still be regarded as a good annotation rate and might even be published. Yet, the fact that we observe such a massive drop in annotation rates must be regarded as a red flag that our machine learning model is overfitting. This red flag shines even brighter if we compare those numbers to the annotation rate of CSI:FingerID without any metascore addition. It turns out that CSI:FingerID reaches 49.6% correct annotation in a structure-disjoint evaluation. Somehow, we forgot to report this important baseline number in the Results section.
In fact, we do not even have to know those numbers from evaluation, to suspect that something fishy is going on. We have trained a neural network with 68.3 k parameters on a training dataset containing 250 queries, corresponding to 250 positive samples (see Methods). Even tricks such as regularization, dropout [
13], or batch normalization [
20]—which we deliberately did not use—could not prevent the resulting model from overfitting to the training data. If the number of training samples is small, then the use of a large “vanilla” deep neural network (also known as multilayer perceptron) should raise some doubt. During the last years, numerous network architectures were developed that substantially reduced the number of parameters in the model, in order to improve its generalization capabilities: Convolutional neural networks for image recognition are a prominent example. Yet, unless there is a convincing reason why such network architecture should be applied to the problem at hand, we cannot simply grab a random architecture and hope that it will improve our results! Stating that a particular model architecture is “state-of-the-art” in machine learning, does not mean it is reasonable to be applied. Machine learning cannot do magic.
For generalizability, less is often more. As reported above, we also trained a sparser geometry of the neural network with only one hidden layer of 12 neurons. This implies that we have substantially fewer parameters in our model. In contrast to our claims in the results section, this does not mean that the model is less powerful. Rather, there is a good chance that the smaller model will perform better on any independent data. We deliberately made it even harder for the small model to “learn”, using 30% dropout when training the network [
13]. This means that 30% of the neurons in the hidden layer are randomly removed before forward and backpropagation is used to update edge weights. Doing so considerably worsened the performance of the model when trained on the evaluation data (memorizing) but simultaneously, improved the performance when trained on the training data and evaluated on the evaluation data (learning). For this more sensible machine learning model, we reach 79.5% correct annotations when training and evaluating on the test dataset, and 76.4% when training on the training data and evaluating on the evaluation data.
Do not evaluate using your training data, reloaded. Unfortunately, the separation of the training and test sets is not as easy as one may think. For an in silico method, saying “we used dataset A for training and dataset B for evaluation” is insufficient. The first thing to notice is that the same spectra are often found in more than one spectral library, and we have to ensure that we do not train on the same data we evaluate on. A prominent example involves the data from the CASMI 2016 contest, which have been repeatedly used for method evaluations. These data were later included in spectral libraries such as GNPS (Global Natural Products Social) and MoNA (MassBank of North America). Hence, a method trained on GNPS and evaluated on CASMI 2016 has seen all the data, and the reported numbers are meaningless for an in silico method.
Yet, even a spectrum-disjoint evaluation is insufficient. If the same compound is measured twice for two libraries, then the resulting spectra will be extremely similar. In fact, library search in public and commercial spectral libraries is based on this observation. Hence, we must ensure that the same compound is never found in training and test data, to avoid that we are basically evaluating spectral library search, which we already know to work rather well. Unfortunately, the rabbit hole goes deeper. This compound-disjoint evaluation is still insufficient. Some compounds (say, L- and D-threose, threose and erythrose) may just differ in their stereochemistry. Yet, stereochemistry has only a minor impact on the fragmentation of a compound. Hence, evaluations of in silico methods ignore stereochemistry and evaluate search results at the level of molecular structures. (At present, an in silico method evaluation claiming to differentiate L/D-threose and L/D-erythrose based on MS/MS data, must not be taken seriously.) To this end, we have to treat all of those compounds as the same structure and ensure that our evaluation is structure-disjoint. These are the minimum requirements for an in silico evaluation to be reasonable.
There are situations that require an even stricter separation of training and test data. For example, the second evaluation of Mad Hatter was a molecular formula-disjoint evaluation, meaning that compounds with the same molecular formula must not appear in training and test data. This implies that the evaluation is also structure-disjoint. The stricter molecular formula-disjoint evaluation is advisable in cases where one suspects that the molecular formula is already giving away too much information to a machine learning model. Finally, scaffold-disjoint evaluations ensure that compounds with the same scaffold must not appear in training and test data; such evaluations are common in virtual screening. From our experience, we can say that a scaffold-disjoint evaluation is not mandatory for in silico methods that analyze MS/MS data. Minor side group changes to a scaffold sometimes strongly affect fragmentation and sometimes not, making it impossible for machine learning to simply memorize spectra. Yet, for methods that consider fragmentation through Electron Ionization (EI), a scaffold-disjoint evaluation is mandatory! Here, each compound is found many times in the library with different types of derivatization, as these derivatization techniques are part of the experimental protocol. If we evaluate the machine learning model on a compound for which ten derivatives are present in the training data, chances are high that the model can spot some similarity to one of the derivatives and basically falls back to memorizing.
Some methods employ machine learning without clearly stating so. Assume that a method adds the scores of two in silico tools. Clearly, we require a multiplier for one of the scores, to make them comparable. Yet, how do we find this multiplier? In fact, it is not too complicated to come up with a set of feature multipliers for Mad Hatter, avoiding the machine learning part of the manuscript. Yet, what we are building—manually or automated—is (highly similar to) a linear classifier from machine learning. Hence, our evaluations must be carried out structure-disjoint (for MS/MS data) or scaffold-disjoint (for EI data). Results that fail to do so are basically meaningless.
5. Metascores
Unlike many publications from the last years, the resulting procedures for training and evaluation of Mad Hatter now follow established standards in machine learning. In fact, we went slightly beyond those standards by using a molecular formula-disjoint evaluation setup. Yet, we have used features that are quite obviously nonsense and should not be used for a metascore. Even less so, they should allow us to annotate compounds at the rates reported above! Recall that in a molecular formula-disjoint evaluation, Mad Hatter reached 76.4% correct annotations when searching PubChem. How is this possible? The thing is, Mad Hatter uses a metascore; our intuition is highly misleading when it comes to metascores.
What is a metascore. It is important to understand that in general, “metascores” have nothing to do with “metadata”, except for the prefix. This appears to be a common misconception. Metadata is data about your experiment. It may be the expected mass accuracy of the measurements, the column type used for chromatography, or the type of sample one is looking at. Such information is already used, to a certain extent, by all existing in silico methods. For example, we always have to tell these methods about the expected mass accuracy of our measurements.
In contrast, a “metascore” is simply integrating two or more different scores into one. For example, we may combine the scores of two or more in silico methods, such as MetFrag and CFM-ID in [
21]. Second, we may combine in silico methods and retention time prediction [
21,
22,
23,
24]. Third, we may combine an in silico method with taxonomic information about the analyzed sample [
25]. Numerous other types of metascores can be imagined; all of them have in common that they rely on data or metadata of the sample we are analyzing. Such metascores are not what we will talk about in the rest of this paper as they do not have the intrinsic issues discussed in the following.
Yet, in the field of in silico methods, the term “metascore” is often used with a different type of method in mind. In many cases, this term describes a method that combines an in silico method and “side information” from the structure databases.
Mad Hatter is an example of such a metascore, as it uses information from the structure database description (
Table 1) to improve its search results. Such metascores do not rely on metadata; instead, they use information that has nothing to do with the sample, the experimental setup, or the data.
In the remainder of this manuscript, we will speak about metascores when in fact, a more precise denomination would be “metascore of the Mad Hatter type”. For readability, we will stick to the shorter term. We remind the reader that our critique only applies to those metascores that use “side information” from the searched structure database.
Blockbuster metabolites. Consider a movie recommendation system that, based on your preferences, suggests new movies to watch. Such recommendation systems are widely-known these days, in particular from streaming services such as Netflix, Disney+, or Amazon Prime. Yet, a recommendation system may completely ignore the user’s preferences and suggest blockbuster movies all the time; it may choose films according to their financial success. Recommendations will sometimes appear very accurate; after all, we all love blockbuster movies, at least statistically speaking. In evaluations, we will not notice that the recommendation system is basically useless. In practice, we will.
Similar to blockbuster movies, we introduce the term “blockbuster metabolite” for a small molecule that is “better” than others. We stress that this term is meant to include compounds of biological interest, including drugs and toxic compounds. How can a compound be “better” than others? Different features have been suggested to differentiate “interesting” from “uninteresting” compounds. This includes citation counts (the number of papers, patents, etc., that contain the compound), production volume, or the number of molecular structure databases a compound is contained in. A metascore will combine the original score that considers the MS/MS data, via some function that also considers, say, the citation count.
A blockbuster metabolite is a compound in the structure database we search in that has a much larger value of the considered feature than all other compounds in the structure database. For example, the compound may have ten thousand citations, whereas most compounds in PubChem have none. In more detail, the compound only needs to have more citations than all compounds it is competing with; in silico methods use the precursor m/z to filter the database for valid candidates, so our compound needs to have a higher citation count than all compounds with a similar mass. The term “blockbuster metabolite” indicates that our metascore may prefer those compounds over all other candidates, independent of the measured MS/MS data.
Reporting metascore annotation rates is futile. Evaluating a metascore method using a spectral library is meaningless and even misleading. In any such evaluation, we report annotation rates on reference data, with the implicit subtext that somewhat similar annotation rates are reached on biological data. Yet, when evaluating a method that is using a metascore, reported annotation rates have no connection whatsoever with rates that you can reach for biological data. This is a general critique of any such evaluation and does not depend on the utilized metascore.
To understand this phenomenon, consider again our Mad Hatter evaluation above. Mad Hatter correctly annotated about 72% of all compounds when we deliberately removed MS/MS data. The same was possible if we replaced our MS/MS spectra with empty spectra or random spectra, resulting in annotation rates of 71.0% and 71.7%, respectively. It is disturbing that an in silico method may basically ignore our data. However, the main discrepancy is that we have evaluated a method using a spectral library full of MS/MS spectra, but then ignored all of the data in our evaluation! We could as well have randomly selected a list of compound masses from a structure database; it is very odd to use compounds from a spectral library, which you then plan to ignore. What is the reason to believe that this evaluation will tell us anything about the performance of a method on biological data? We argue that there is no such reason; in fact, there are convincing arguments why we should not expect similar performances for biological data, be it from metabolomics or environmental research.
Now, why is it not a good idea to evaluate a metascore on a spectral library, and what can we actually learn from such an evaluation? We can learn that the distribution of compounds in any MS/MS library differs substantially from the distribution of compounds of biological interest, or PubChem compounds. Furthermore, we may learn that it is not very complicated to utilize these differences, to make a metascore shine in evaluation. Yet, the actual number that we record as an “annotation rate”, has no meaning.
For the sake of simplicity, let us focus on citation counts, a feature that has repeatedly been suggested and used for metascores. Similar arguments hold for other features.
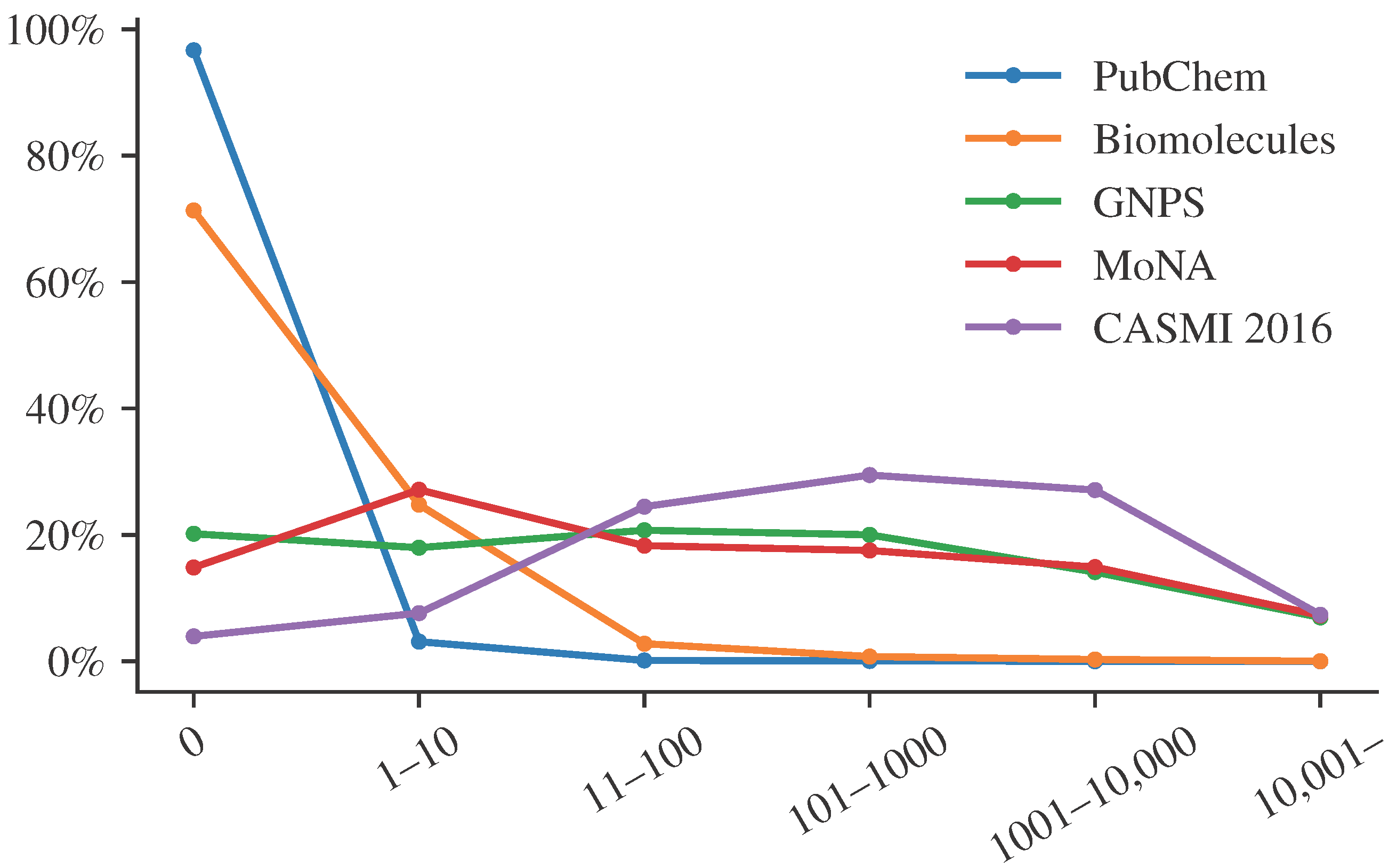
Figure 1 shows the distribution of citation counts for different structure databases and libraries. We see that citation counts differ substantially between PubChem, a database containing “molecules of biological interest”, and spectral libraries. We see that most compounds from PubChem have very few citations, whereas biomolecules have larger citation counts. Notably, these are relative abundances. Every compound from the biomolecular structure databases is also found in PubChem. Yet, the vast majority of compounds in PubChem are not in the biomolecular structure database, and these compounds usually have very low citation counts. The important point here is that compounds found in a spectral library have many more citations than the average biomolecule, and even more so, than the average PubChem molecule. The reasons for that go in both directions. On the one hand, a compound that has many citations is more likely to be included in a spectral library. On the other hand, a compound that is included in a spectral library will be annotated more often in biological samples and, hence, has a much higher chance to be mentioned in a publication. These are just two reasons, we assume there are many more.
Clearly, there may be compounds in a spectral library that are not blockbuster metabolites. For example, CASMI 2016 contains MS/MS data for 4-[(4-hydroxynaphthalen-1-yl)diazenyl]benzenesulfonic acid, which has only a single citation. These compounds may be wrongly annotated by a metascore as soon as we find a blockbuster metabolite with the same mass. However, statistically speaking, such cases are the exception and not the rule. The vast majority of compounds in the CASMI 2016 spectral library are blockbuster metabolites, and using a metascore strongly improves our evaluation results.
Unfortunately, even blind competitions such as CASMI cannot avoid this issue, except by explicitly banning the use of metascores. In CASMI 2016, category #3 explicitly allowed metascores such as Mad Hatter. This category was termed “best automatic structural identification—full information”. It explicitly stated that “any form of additional information can be used”, and mentions patents and reference counts as examples. The best submission in this category reached 70% correct annotations. Throughout this paper, we only used information also available to us during the CASMI 2016 contest. Hence, Mad Hatter could have participated in this category, reaching 76% correct annotations and winning the competition. CASMI is a blind competition, and nobody would have even known about our lunatic features.
Notably, not only the evaluation of a metascore method is futile. Also, using annotations from a metascore method to derive statistics about an experimental sample is futile and even misleading. As discussed, we mostly annotate blockbuster metabolites and miss out on all other compounds present in the sample. Hence, mapping those annotations to metabolic pathways or compound classes to, say, check for expression change, is not advisable. Doing so will tell us something about blockbuster metabolites and how they map to pathways and compound classes, but very little (if anything) about the data.
A method shining in evaluations does not have to be reasonable. We have deliberately chosen outrageous features for Mad Hatter. Other metascores often use “reasonable” features such as the number of citations, or the number of biological databases a molecular structure is contained in. The reasoning behind this appears to be as follows. If a metascore sounds reasonable and performs well in evaluations, we can safely infer that the approach is justified. Showing that you can build a “good” metascore using outrageous features disproves this implicit reasoning. Mad Hatter works great in evaluations, but it is definitely not a reasonable method. Hence, you must never assume that a method is reasonable or justified simply because it works well in evaluations. A metascore that uses “reasonable” features may or may not work in evaluations; however, we cannot infer that the resulting method is reasonable, based on this fact.
Let us take a look behind the curtain. Why is Mad Hatter capable to distinguish between blockbuster and other compounds, despite the fact that the Mad Hatter features are clearly nonsense? For certain features, it is not hard to see why these features favor blockbuster metabolites. For example, blockbusters usually possess a shorter synonym, such as “aspirin” instead of “acetylsalicylic acid” or “2-(Acetyloxy)benzoic acid”. This short synonym is then used as the compound name; consequently, the length of the compound name contains viable information on whether a compound is a blockbuster or not. Another such feature is the number of vowels in the shelf life description. A long description points toward a blockbuster metabolite; a long description also contains more vowels.
Potentially more dangerous are features for which, even at second thought, we cannot find a reasonable explanation of what is going on. Consider the highly counterintuitive melting point feature. This value is present only for compounds that have been analyzed in some labs and is left empty for all others. If we had filled in missing values with some reasonable value (say, the average temperature over all compounds where the value is provided), then this feature would carry little useful information for Mad Hatter. Yet, we have taken the liberty to fill in missing values in the worst possible way: We used −273.15 °C for those compounds where no melting point was given. This is extremely far off from all compounds that have an entry; hence, this nonchalant little “trick” makes it trivial for machine learning to sort out “uninteresting compounds” where no melting point is provided. Luckily for us, those “uninteresting compounds” are also (almost) never contained in any spectral library. We have hidden the details on missing values somewhere in the methods section, making it hard for a reader to grasp what is going on. We have done this intentionally; yet, we also could have done so out of inexperience with machine learning and how to properly describe it. Recording the temperature in Kelvin plus filling missing values with zeroes is not so far-fetched.
Finally, some features are merely red herrings (say, record number modulo 42, structure complexity); we expect that removing them will not have a large impact on the performance of the metascore, if any. Yet, including them will not impair machine learning, either. Be warned that some of the red herring features may be picked up by machine learning for filtering candidates, in a way unpredictable and possibly incomprehensible for the human brain.
Any type of metascore will, to a varying degree, allow us to annotate blockbuster metabolites and only blockbuster metabolites. It does not really matter if our features are “reasonable” or “unreasonable”; machine learning will find its way to deduce “helpful information” from the data.
Other compounds become invisible. Blockbuster metabolites are somewhat similar to what is sometimes said about Hollywood stars. They need all the attention, and they cannot bear rivaling compounds. In detail, blockbuster metabolites will often overshadow other metabolites with the same mass. Irrespective of the actual MS/MS query spectrum that we use for searching, the metascore method will return the blockbuster metabolite. All compounds overshadowed by a blockbuster metabolite become basically invisible. For examples of where this goes wrong, see
Table 2.
Notably, an invisible compound may not appear invisible at first glance. For example, “ethyl 4-dimethylaminobenzoate” (74 citations) was incorrectly annotated as “3,4-methylenedioxymethamphetamine” (4.5k citations, also known as “ecstasy”) in our
Mad Hatter evaluation. Clearly,
Mad Hatter does not consider citation counts but rather the number of words starting with ‘U’ (153 vs. 377) and the number of “was it a cat I saw?” (136 vs. 420); the result is the same. See
Table 2 (left). As noted, blockbuster metabolites are similar to divas, and the biggest diva takes it all.
Unfortunately, overshadowing is not only an issue when reporting annotation statistics. Assume that you want to annotate methyl 2-pyrazinecarboxylate from
Table 2 (right) based on the measured query spectrum. Good news: CSI:FingerID was actually able to annotate the correct compound structure. Bad news: If you are using a metascore, you will never get to know. The metascore replaced the correct answer with an incorrect one (namely, 4-nitroaniline), as the incorrect one was more often cited. Again, it is not citations that
Mad Hatter considers, but the result is the same. If we order compound candidates by CSI:FingerID score, we may want to manually correct annotations every now and then, based on citation counts and expert knowledge. Yet, if we only consider the metascore, there is no way for us to spot situations where CSI:FingerID is correct and citation counts are misleading.
The above introduces yet another issue. We might believe that we are searching PubChem when in truth, we are searching in a much smaller database. Using a metascore, we can basically drop all invisible metabolites from the searched structure database without ever changing the search results, regardless of the actual MS/MS query spectrum. Unfortunately, we do not know in which database we are actually searching and, hence, we cannot report this information. This is no good news for reproducibility or good scientific practice. As an example not from CASMI 2016, consider molecular formula C17H21NO4. PubChem contains 8384 compounds with this molecular formula. Cocaine (PubChem ID 446220) has 35,200 citations, many more than any other compound. It is nevertheless unreasonable to assume that every compound in every sample with this molecular formula is cocaine. Cocaine may even mask other highly cited compounds, such as scopolamine (PCID 3000322, 8.9 k citations) or fenoterol (PCID 3343, 1.9 k citations). However, most importantly, more than 8100 structures in PubChem with this molecular formula do not have a single citation and will be practically invisible to the metascore.
Clearly, there are queries for which overshadowing is not an issue. In particular, there are no blockbusters in the database for certain precursor masses. In these cases, the metascore method will behave similar to the original method without a metascore. Yet, this does not disprove the above reasoning: For a large fraction of queries, and particularly for biological queries, blockbusters will overshadow other compounds.
Metabolomics vs. environmental research. So far, we have concentrated on metabolomics and biological data in our argumentation. What about environmental research? There, we have highly reasonable prior information stored in the structure databases. Namely, the production volumes of compounds. The rationale behind this is that something that is not produced by mankind, should also not be annotated in an environmental sample. This appears to be very reasonable, but unfortunately, most of the arguments formulated above remain valid. Clearly, this is true for our argumentation regarding the evaluation setup, discussed in the previous section. However, also, with regard to blockbuster compounds, we find that most arguments still hold. An evaluation of a metascore using a spectral library is futile; production rates are a sensible feature to use, but that does not imply that the resulting metascore is sensible; compounds can mask other compounds with lower production volume; excellent hits can be masked by the metascore; we do not really need MS/MS data but instead, could simply use the monoisotopic mass plus the metascore; we cannot report the structure database we are searching in; and so on.
Targeted vs. untargeted analysis, and the right to refuse to annotate. Studies that employ LC–MS or LC–MS/MS for compound annotation are often referred to as “untargeted” by the authors. This is done to differentiate the instrumental setup from, say, multiple reaction monitoring experiments where we have to choose the compounds to be annotated before we start recording data. Is this nomenclature reasonable? The experimental setup is indeed not targeted to a specific set of compounds.
We argue that doing so is at least misleading, as it mixes up the employed instrumental technology, and the actual experiment we are conducting. Data analysis must be seen as an integral part of any experiment. If we use LC–MS/MS to measure our data, but then limit ourselves to a single compound that we annotate, then this is not an untargeted experiment. Our data would allow us to go untargeted, but if we do not do that, we also should not call it by this name. If we go from one to a thousand compounds, we are still targeting exactly those 1000 compounds. Metaphorically speaking, if you build a propeller engine into a car, you will not fly.
Spectral library search is a “mostly untargeted” data analysis as we restrict ourselves to compounds present in a library, but do not impose further prior knowledge. Any compound in the library has the same chance of getting annotated, as long as it fits the data. A massive advantage in comparison to the “list of masses” is that we reserve the right not to annotate certain compounds in the dataset. If the query spectra do not match anything from the spectral library, then we can label those “unknowns”. We are aware that such “unknown” labels are generally considered a pain in metabolomics research [
30]. Yet, knowing that we cannot annotate a certain metabolite is definitely better than assigning a bogus annotation!
In silico methods are a logical next step for untargeted data analysis, as we extend our search to all compounds in a more comprehensive molecular structure database. Yet, even the structure database is incomplete and we might miss the correct structure. Certain methods try to overcome these restrictions altogether [
31,
32,
33,
34,
35,
36], and we expect this to be an active field of research in the years to come.
Using a metascore forces our in silico method back into the targeted corset, as invisible compounds can never be found by the metascore. In a way, metascore methods may even be considered “more targeted” than spectral library search, as the latter does not give different priors to compounds. Metascores are, therefore, in fundamental opposition to the central paradigm behind in silico methods. These methods were developed to overcome the issue of incomplete spectral libraries and, hence, to make data analysis “less targeted”. It is a puzzling idea to go more and less targeted at the same time. In addition, we mostly lose the possibility to flag certain compounds as “unknowns”, if we search a large database such as PubChem. As we have argued above, the knowledge of what we can and what we cannot confidently annotate is actually very useful [
37].








{kind=link}
{kind=link}
