

Hyperthyroidism and Wnt Signaling Pathway: Influence on Bone Remodeling

 ,
,
Abstract
1. Introduction
2. Graves’ Disease
3. Clinical Symptoms and Treatment of GD
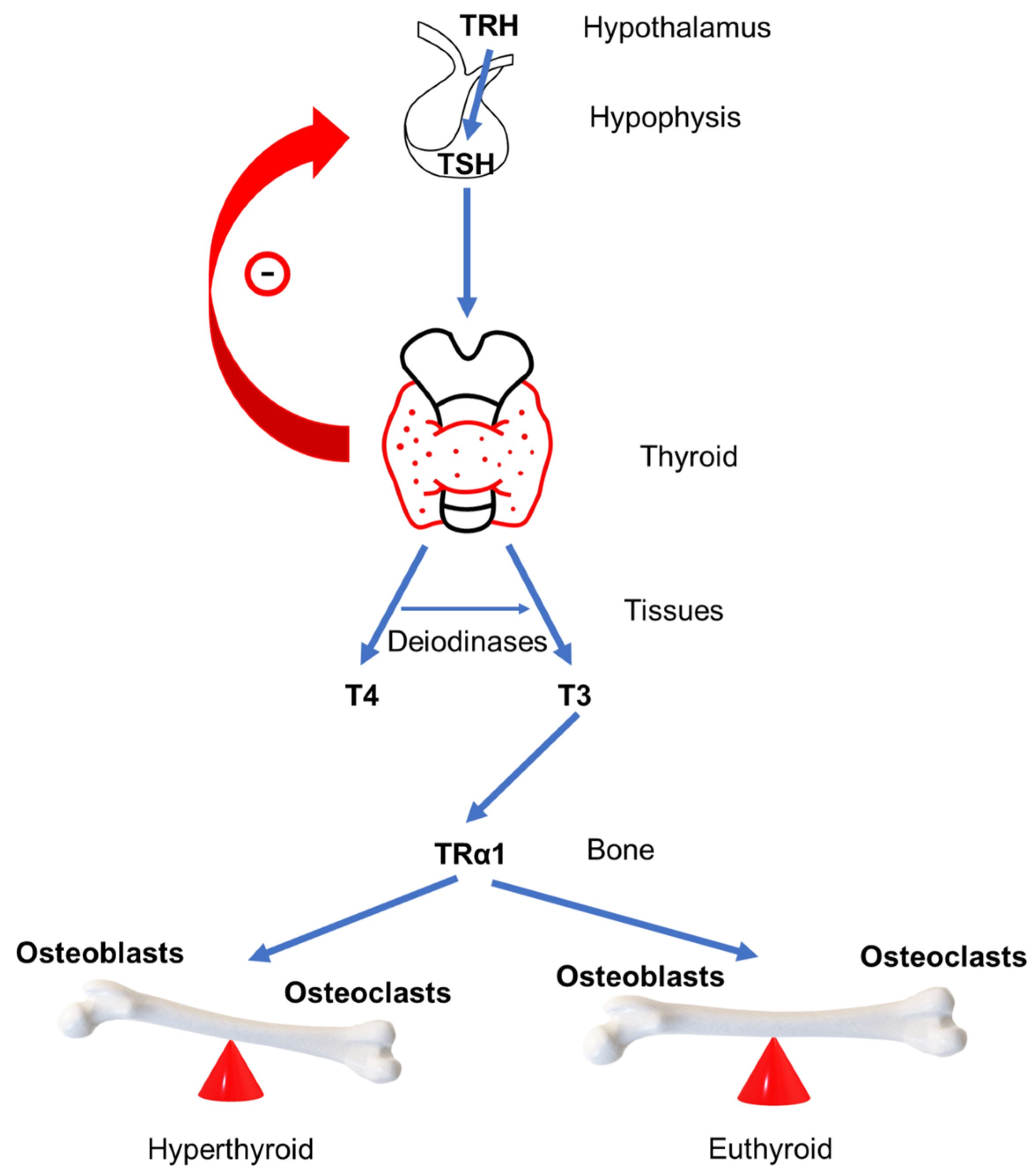
4. GD Impact on Bone Remodeling
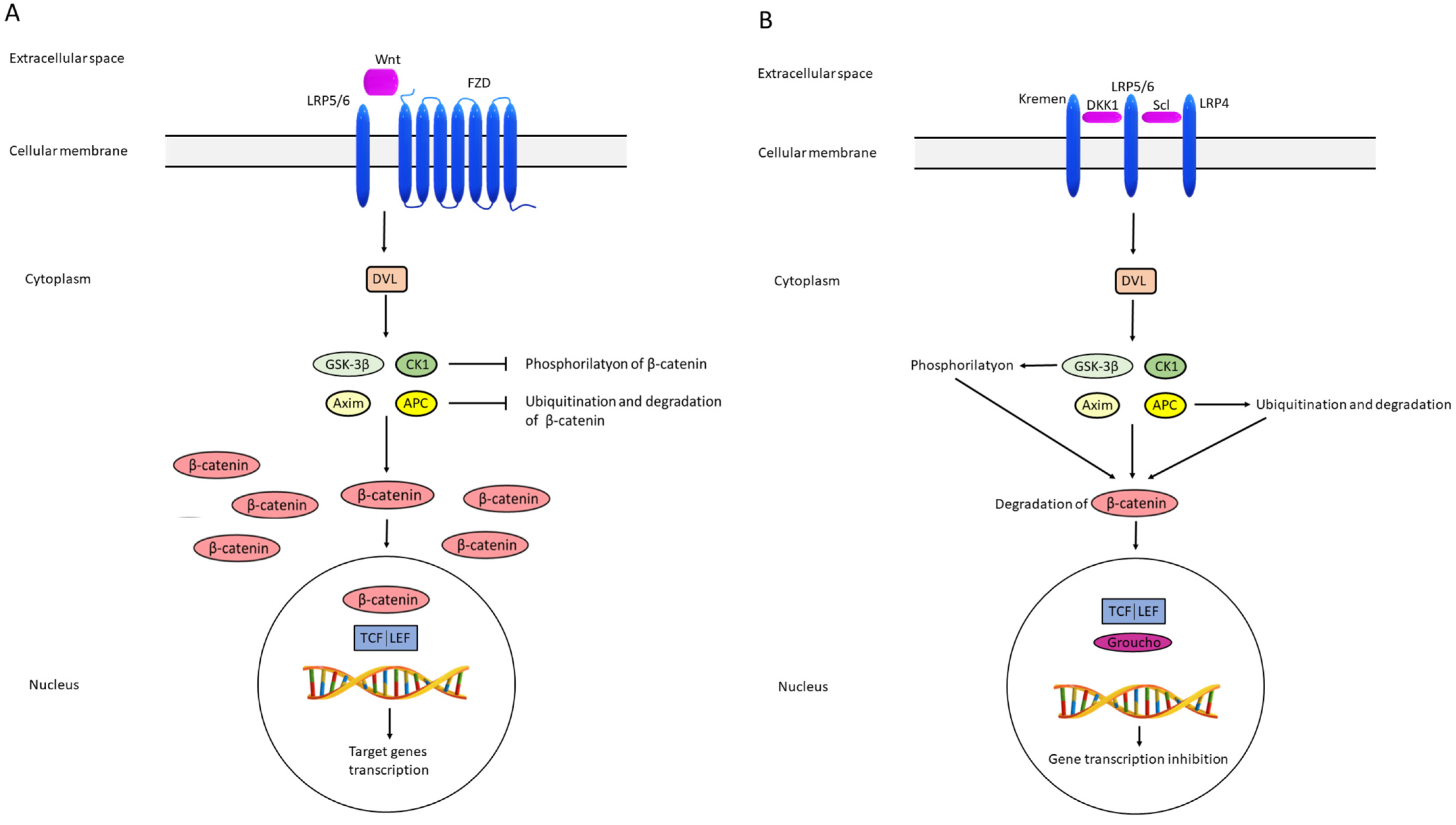
5. Wnt/β-Catenin Pathway
6. Wnt/β-Catenin Signaling Pathway and Thyroid Hormones

{kind=link}
{kind=link}
| Author | Study Design | Hyperthyroidism Etiology | Analyzed Wnt Inhibitor | Main Results |
|---|---|---|---|---|
| Tsourdi 2015 [71] | Animal models | Induced hyperthyroidism | Sclerostin, DKK1 | Increased sclerostin and decreased DKK1 serum concentrations in hyperthyroid mice; |
| Tsourdi 2019 [73] | Animal models | Induced hyperthyroidism | DKK1 | Loss of DKK1 is not sufficient to fully reverse thyroid hormone– induced changes in bone mass and bone turnover |
| Skowrońska-Jóźwiak 2012 [68] | In 15 patients, sclerostin was measured at diagnosis of hyperthyroidism and after 6–10 weeks of treatment with thiamazole | Graves’ disease or toxic multinodular goitre | Sclerostin | A significant decrease in serum sclerostin levels after achieving euthyroid state |
| Skowrońska-Jóźwiak 2015 [69] | In 33 patients, serum sclerostin was measured at diagnosis of hyperthyroidism and after 6–10 weeks of treatment with thiamazole | Graves’ disease or toxic multinodular goitre | Sclerostin | After treatment of hyperthyroidism, a significant decrease in serum sclerostin was measured. |
| Sarıtekin 2017 [72] | 24 patients with hyperthyroidism, yet untreated and 24 voluntary normal persons | Multinodular goiter and Graves’ disease | Sclerostin | There was no difference in the serum sclerostin levels between the hyperthyroid group and the control group. |
| Mihaljević 2020 [70] | An amount of 30 patients with hypothyroidism, hyperthyroidism, and subclinical hyperthyroidism, as well as 10 euthyroid controls | Graves’ disease and subclinical hyperthyroidism due to thyroxine suppressive therapy | Sclerostin | Sclerostin levels were significantly elevated in hyperthyroidism compared to subclinical hyperthyroidism and control group |
| Mudri 2022 [32] | Longitudinal study included 37 patients in which serum sclerostin and DKK1 were measured at diagnosis of GD and after one year of ATD therapy | Graves’ disease | Sclerostin, DKK1 | Significant decrease in serum sclerostin and significant increase in serum DKK1 in euthyroid state after one year |
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chazenbalk, G.D.; Pichurin, P.; Chen, C.R.; Latrofa, F.; Johnstone, A.P.; McLachlan, S.M.; Rapoport, B. Thyroid-stimulating autoantibodies in Graves disease preferentially recognize the free A subunit, not the thyrotropin holoreceptor. J. Clin. Investig. 2002, 110, 209–217. [Google Scholar] [CrossRef]
- Antonelli, A.; Ferrari, S.M.; Ragusa, F.; Elia, G.; Paparo, S.R.; Ruffilli, I.; Patrizio, A.; Giusti, C.; Gonnella, D.; Cristaudo, A.; et al. Graves’ disease: Epidemiology, genetic and environmental risk factors and viruses. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101387. [Google Scholar] [CrossRef]
- Nicolaisen, P.; Obling, M.L.; Winther, K.H.; Hansen, S.; Hermann, A.P.; Hegedüs, L.; Bonnema, S.J.; Brix, T.H. Consequences of Hyperthyroidism and Its Treatment for Bone Microarchitecture Assessed by High-Resolution Peripheral Quantitative Computed Tomography. Thyroid 2021, 31, 208–216. [Google Scholar] [CrossRef]
- Vestergaard, P.; Mosekilde, L. Hyperthyroidism, bone mineral, and fracture risk--a meta-analysis. Thyroid 2003, 13, 585–593. [Google Scholar] [CrossRef]
- Lin, C.; Jiang, X.; Dai, Z.; Guo, X.; Weng, T.; Wang, J.; Li, Y.; Feng, G.; Gao, X.; He, L. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J. Bone Miner. Res. 2009, 24, 1651–1661. [Google Scholar] [CrossRef]
- Qiang, Y.W.; Barlogie, B.; Rudikoff, S.; Shaughnessy, J.D. Dkk1-induced inhibition of Wnt signaling in osteoblast differentiation is an underlying mechanism of bone loss in multiple myeloma. Bone 2008, 42, 669–680. [Google Scholar] [CrossRef]
- Li, J.; Sarosi, I.; Cattley, R.C.; Pretorius, J.; Asuncion, F.; Grisanti, M.; Morony, S.; Adamu, S.; Geng, Z.; Qiu, W.; et al. Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone 2006, 39, 754–766. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Sato, A.Y.; Bellido, T. Role and mechanism of action of sclerostin in bone. Bone 2017, 96, 29–37. [Google Scholar] [CrossRef]
- Kim, J.H.; Liu, X.; Wang, J.; Chen, X.; Zhang, H.; Kim, S.H.; Cui, J.; Li, R.; Zhang, W.; Kong, Y.; et al. Wnt signaling in bone formation and its therapeutic potential for bone diseases. Ther. Adv. Musculoskelet. Dis. 2013, 5, 13–31. [Google Scholar] [CrossRef]
- Smith, T.J.; Hegedüs, L. Graves’ Disease. N. Engl. J. Med. 2016, 375, 1552–1565. [Google Scholar] [CrossRef]
- Bartalena, L. Diagnosis and management of Graves disease: A global overview. Nat. Rev. Endocrinol. 2013, 9, 724–734. [Google Scholar] [CrossRef]
- Chi, H.C.; Tsai, C.Y.; Tsai, M.M.; Yeh, C.T.; Lin, K.H. Molecular functions and clinical impact of thyroid hormone-triggered autophagy in liver-related diseases. J. Biomed. Sci. 2019, 26, 24. [Google Scholar] [CrossRef]
- Mancino, G.; Miro, C.; Di Cicco, E.; Dentice, M. Thyroid hormone action in epidermal development and homeostasis and its implications in the pathophysiology of the skin. J. Endocrinol. Investig. 2021, 44, 1571–1579. [Google Scholar] [CrossRef]
- Yen, P.M. Physiological and molecular basis of thyroid hormone action. Physiol. Rev. 2001, 81, 1097–1142. [Google Scholar] [CrossRef]
- Delitala, A.P.; Scuteri, A.; Doria, C. Thyroid Hormone Diseases and Osteoporosis. J. Clin. Med. 2020, 9, 1034. [Google Scholar] [CrossRef]
- Welsh, K.J.; Soldin, S.J. Diagnosis of endocrine disease: How reliable are free thyroid and total T3 hormone assays? Eur. J. Endocrinol. 2016, 175, R255–R263. [Google Scholar] [CrossRef]
- Feldt-Rasmussen, U.; Krogh Rasmussen, Å. Thyroid Hormone Transport and Actions. Pediatr. Adolesc. Med. 2007, 11, 80–103. [Google Scholar] [CrossRef]
- Friesema, E.C.; Jansen, J.; Visser, T.J. Thyroid hormone transporters. Biochem. Soc. Trans. 2005, 33, 228–232. [Google Scholar] [CrossRef]
- Cardoso, L.F.; Maciel, L.M.; Paula, F.J. The multiple effects of thyroid disorders on bone and mineral metabolism. Arq. Bras. Endocrinol. Metabol. 2014, 58, 452–463. [Google Scholar] [CrossRef]
- Bassett, J.H.; Williams, G.R. Role of Thyroid Hormones in Skeletal Development and Bone Maintenance. Endocr. Rev. 2016, 37, 135–187. [Google Scholar] [CrossRef]
- Bianco, A.C.; Kim, B.W. Deiodinases: Implications of the local control of thyroid hormone action. J. Clin. Investig. 2006, 116, 2571–2579. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef]
- Nicholls, J.J.; Brassill, M.J.; Williams, G.R.; Bassett, J.H. The skeletal consequences of thyrotoxicosis. J. Endocrinol. 2012, 213, 209–221. [Google Scholar] [CrossRef]
- Längericht, J.; Krämer, I.; Kahaly, G.J. Glucocorticoids in Graves’ orbitopathy: Mechanisms of action and clinical application. Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018820958335. [Google Scholar] [CrossRef]
- Chin, Y.H.; Ng, C.H.; Lee, M.H.; Koh, J.W.H.; Kiew, J.; Yang, S.P.; Sundar, G.; Khoo, C.M. Prevalence of thyroid eye disease in Graves’ disease: A meta-analysis and systematic review. Clin. Endocrinol. 2020, 93, 363–374. [Google Scholar] [CrossRef]
- Subekti, I.; Pramono, L.A. Current Diagnosis and Management of Graves’ Disease. Acta Med. Indones 2018, 50, 177–182. [Google Scholar]
- Burch, H.B.; Cooper, D.S. Management of Graves Disease: A Review. JAMA 2015, 314, 2544–2554. [Google Scholar] [CrossRef]
- Zoričić-Cvek, S.A.; Bobinac, D.; Đudarić, L.; Cvijanović, O. The remodeling of the skeleton. Med. Flumensis 2015, 51, 11. [Google Scholar]
- Kenkre, J.S.; Bassett, J. The bone remodelling cycle. Ann. Clin. Biochem. 2018, 55, 308–327. [Google Scholar] [CrossRef]
- Williams, G.R. Thyroid hormone actions in cartilage and bone. Eur. Thyroid J. 2013, 2, 3–13. [Google Scholar] [CrossRef]
- Gorka, J.; Taylor-Gjevre, R.M.; Arnason, T. Metabolic and clinical consequences of hyperthyroidism on bone density. Int. J. Endocrinol. 2013, 2013, 638727. [Google Scholar] [CrossRef]
- Mudri, D.; Kizivat, T.; Mihaljević, I.; Ćurčić, I.B. Wnt Inhibitors and Bone Mineral Density in Patients with Graves’ Disease Treated with Antithyroid Drugs: A Preliminary Prospective Study. Metabolites 2022, 12, 711. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- Skah, S.; Uchuya-Castillo, J.; Sirakov, M.; Plateroti, M. The thyroid hormone nuclear receptors and the Wnt/β-catenin pathway: An intriguing liaison. Dev. Biol. 2017, 422, 71–82. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Z.; Tang, Y.; Xiao, Q. The involvement of noncanonical Wnt signaling in cancers. Biomed. Pharmacother. 2021, 133, 110946. [Google Scholar] [CrossRef]
- Lademann, F.; Tsourdi, E.; Hofbauer, L.C.; Rauner, M. Thyroid Hormone Actions and Bone Remodeling—The Role of the Wnt Signaling Pathway. Exp. Clin. Endocrinol. Diabetes 2020, 128, 450–454. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target Ther. 2022, 7, 3. [Google Scholar] [CrossRef]
- Flack, J.E.; Mieszczanek, J.; Novcic, N.; Bienz, M. Wnt-Dependent Inactivation of the Groucho/TLE Co-repressor by the HECT E3 Ubiquitin Ligase Hyd/UBR5. Mol. Cell 2017, 67, 181–193.e185. [Google Scholar] [CrossRef]
- Krishnan, V.; Bryant, H.U.; Macdougald, O.A. Regulation of bone mass by Wnt signaling. J. Clin. Investig. 2006, 116, 1202–1209. [Google Scholar] [CrossRef]
- Zheng, J.; Maerz, W.; Gergei, I.; Kleber, M.; Drechsler, C.; Wanner, C.; Brandenburg, V.; Reppe, S.; Gautvik, K.M.; Medina-Gomez, C.; et al. Mendelian Randomization Analysis Reveals a Causal Influence of Circulating Sclerostin Levels on Bone Mineral Density and Fractures. J. Bone Miner. Res. 2019, 34, 1824–1836. [Google Scholar] [CrossRef]
- Ke, H.Z.; Richards, W.G.; Li, X.; Ominsky, M.S. Sclerostin and Dickkopf-1 as therapeutic targets in bone diseases. Endocr. Rev. 2012, 33, 747–783. [Google Scholar] [CrossRef]
- Carrillo-López, N.; Martínez-Arias, L.; Fernández-Villabrille, S.; Ruiz-Torres, M.P.; Dusso, A.; Cannata-Andía, J.B.; Naves-Díaz, M.; Panizo, S.; On behalf of the European Renal Osteodystrophy (EUROD) Workgroup. Role of the RANK/RANKL/OPG and Wnt/β-Catenin Systems in CKD Bone and Cardiovascular Disorders. Calcif. Tissue Int. 2021, 108, 439–451. [Google Scholar] [CrossRef]
- Kubota, T.; Michigami, T.; Ozono, K. Wnt signaling in bone metabolism. J. Bone Miner. Metab. 2009, 27, 265–271. [Google Scholar] [CrossRef]
- Shan, Y.; Wang, L.; Li, G.; Shen, G.; Zhang, P.; Xu, Y. Methylation of bone. Biochem. Cell Biol. 2019, 97, 369–374. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Bellido, T. Osteocytes and Skeletal Pathophysiology. Curr. Mol. Biol. Rep. 2015, 1, 157–167. [Google Scholar] [CrossRef]
- Schaffler, M.B.; Kennedy, O.D. Osteocyte signaling in bone. Curr. Osteoporos. Rep. 2012, 10, 118–125. [Google Scholar] [CrossRef]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The osteocyte: An endocrine cell… and more. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef]
- Sebastian, A.; Loots, G.G. Transcriptional control of Sost in bone. Bone 2017, 96, 76–84. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, M.; Xu, S.; Li, S.; Yang, M.; Su, T.; Yuan, Z.; Peng, H. The Clinical Significance of Dickkopf Wnt Signaling Pathway Inhibitor Gene Family in Head and Neck Squamous Cell Carcinoma. Med. Sci. Monit. 2020, 26, e927368. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, L.; Liu, A. Dickkopf-1: Current knowledge and related diseases. Life Sci. 2018, 209, 249–254. [Google Scholar] [CrossRef]
- Evenepoel, P.; D’Haese, P.; Brandenburg, V. Sclerostin and DKK1: New players in renal bone and vascular disease. Kidney Int. 2015, 88, 235–240. [Google Scholar] [CrossRef]
- Rachner, T.D.; Göbel, A.; Benad-Mehner, P.; Hofbauer, L.C.; Rauner, M. Dickkopf-1 as a mediator and novel target in malignant bone disease. Cancer Lett. 2014, 346, 172–177. [Google Scholar] [CrossRef]
- Paik, J.; Scott, L.J. Romosozumab: A Review in Postmenopausal Osteoporosis. Drugs Aging 2020, 37, 845–855. [Google Scholar] [CrossRef]
- Dentice, M.; Marsili, A.; Zavacki, A.; Larsen, P.R.; Salvatore, D. The deiodinases and the control of intracellular thyroid hormone signaling during cellular differentiation. Biochim. Biophys. Acta 2013, 1830, 3937–3945. [Google Scholar] [CrossRef]
- Sirakov, M.; Skah, S.; Nadjar, J.; Plateroti, M. Thyroid hormone’s action on progenitor/stem cell biology: New challenge for a classic hormone? Biochim. Biophys. Acta 2013, 1830, 3917–3927. [Google Scholar] [CrossRef]
- Natsume, H.; Sasaki, S.; Kitagawa, M.; Kashiwabara, Y.; Matsushita, A.; Nakano, K.; Nishiyama, K.; Nagayama, K.; Misawa, H.; Masuda, H.; et al. Beta-catenin/Tcf-1-mediated transactivation of cyclin D1 promoter is negatively regulated by thyroid hormone. Biochem. Biophys. Res. Commun. 2003, 309, 408–413. [Google Scholar] [CrossRef]
- Guigon, C.J.; Zhao, L.; Lu, C.; Willingham, M.C.; Cheng, S.Y. Regulation of beta-catenin by a novel nongenomic action of thyroid hormone beta receptor. Mol. Cell Biol. 2008, 28, 4598–4608. [Google Scholar] [CrossRef]
- Dentice, M.; Luongo, C.; Ambrosio, R.; Sibilio, A.; Casillo, A.; Iaccarino, A.; Troncone, G.; Fenzi, G.; Larsen, P.R.; Salvatore, D. β-Catenin regulates deiodinase levels and thyroid hormone signaling in colon cancer cells. Gastroenterology 2012, 143, 1037–1047. [Google Scholar] [CrossRef]
- Ely, K.A.; Bischoff, L.A.; Weiss, V.L. Wnt Signaling in Thyroid Homeostasis and Carcinogenesis. Genes 2018, 9, 204. [Google Scholar] [CrossRef]
- Wang, L.; Shao, Y.Y.; Ballock, R.T. Thyroid hormone interacts with the Wnt/beta-catenin signaling pathway in the terminal differentiation of growth plate chondrocytes. J. Bone Miner. Res. 2007, 22, 1988–1995. [Google Scholar] [CrossRef]
- Wang, L.; Shao, Y.Y.; Ballock, R.T. Carboxypeptidase Z (CPZ) links thyroid hormone and Wnt signaling pathways in growth plate chondrocytes. J. Bone Miner. Res. 2009, 24, 265–273. [Google Scholar] [CrossRef]
- Wang, L.; Shao, Y.Y.; Ballock, R.T. Thyroid hormone-mediated growth and differentiation of growth plate chondrocytes involves IGF-1 modulation of beta-catenin signaling. J. Bone Miner. Res. 2010, 25, 1138–1146. [Google Scholar] [CrossRef]
- Furuya, F.; Hanover, J.A.; Cheng, S.Y. Activation of phosphatidylinositol 3-kinase signaling by a mutant thyroid hormone beta receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 1780–1785. [Google Scholar] [CrossRef]
- O’Shea, P.J.; Kim, D.W.; Logan, J.G.; Davis, S.; Walker, R.L.; Meltzer, P.S.; Cheng, S.Y.; Williams, G.R. Advanced bone formation in mice with a dominant-negative mutation in the thyroid hormone receptor β gene due to activation of Wnt/β-catenin protein signaling. J. Biol. Chem. 2012, 287, 17812–17822. [Google Scholar] [CrossRef]
- Skowrońska-Jóźwiak, E.; Krawczyk-Rusiecka, K.; Lewandowski, K.C.; Adamczewski, Z.; Lewiński, A. Successful treatment of thyrotoxicosis is accompanied by a decrease in serum sclerostin levels. Thyroid Res. 2012, 5, 14. [Google Scholar] [CrossRef]
- Skowrońska-Jóźwiak, E.; Lewandowski, K.C.; Adamczewski, Z.; Krawczyk-Rusiecka, K.; Lewiński, A. Mechanisms of Normalisation of Bone Metabolism during Recovery from Hyperthyroidism: Potential Role for Sclerostin and Parathyroid Hormone. Int. J. Endocrinol. 2015, 2015, 948384. [Google Scholar] [CrossRef]
- Mihaljević, O.; Živančević-Simonović, S.; Lučić-Tomić, A.; Živković, I.; Minić, R.; Mijatović-Teodorović, L.; Jovanović, Z.; Anđelković, M.; Stanojević-Pirković, M. The association of circulating sclerostin level with markers of bone metabolism in patients with thyroid dysfunction. J. Med. Biochem. 2020, 39, 436–443. [Google Scholar] [CrossRef]
- Tsourdi, E.; Rijntjes, E.; Köhrle, J.; Hofbauer, L.C.; Rauner, M. Hyperthyroidism and Hypothyroidism in Male Mice and Their Effects on Bone Mass, Bone Turnover, and the Wnt Inhibitors Sclerostin and Dickkopf-1. Endocrinology 2015, 156, 3517–3527. [Google Scholar] [CrossRef]
- Sarıtekin, İ.; Açıkgöz, Ş.; Bayraktaroğlu, T.; Kuzu, F.; Can, M.; Güven, B.; Mungan, G.; Büyükuysal, Ç.; Sarıkaya, S. Sclerostin and bone metabolism markers in hyperthyroidism before treatment and interrelations between them. Acta Biochim. Pol. 2017, 64, 597–602. [Google Scholar] [CrossRef]
- Tsourdi, E.; Colditz, J.; Lademann, F.; Rijntjes, E.; Köhrle, J.; Niehrs, C.; Hofbauer, L.C.; Rauner, M. The Role of Dickkopf-1 in Thyroid Hormone-Induced Changes of Bone Remodeling in Male Mice. Endocrinology 2019, 160, 664–674. [Google Scholar] [CrossRef]
- Rozenberg, S.; Bruyère, O.; Bergmann, P.; Cavalier, E.; Gielen, E.; Goemaere, S.; Kaufman, J.M.; Lapauw, B.; Laurent, M.R.; De Schepper, J.; et al. How to manage osteoporosis before the age of 50. Maturitas 2020, 138, 14–25. [Google Scholar] [CrossRef]
- Majima, T.; Komatsu, Y.; Doi, K.; Takagi, C.; Shigemoto, M.; Fukao, A.; Morimoto, T.; Corners, J.; Nakao, K. Negative correlation between bone mineral density and TSH receptor antibodies in male patients with untreated Graves’ disease. Osteoporos. Int. 2006, 17, 1103–1110. [Google Scholar] [CrossRef]
- Boonya-Ussadorn, T.; Punkaew, B.; Sriassawaamorn, N. A comparative study of bone mineral density between premenopausal women with hyperthyroidism and healthy premenopausal women. J. Med. Assoc. Thai 2010, 93, S1–S5. [Google Scholar]
- Tuchendler, D.; Bolanowski, M. Assessment of bone metabolism in premenopausal females with hyperthyroidism and hypothyroidism. Endokrynol. Pol. 2013, 64, 40–44. [Google Scholar]
- Barbosa, A.P.; Rui Mascarenhas, M.; Silva, C.F.; Távora, I.; Bicho, M.; do Carmo, I.; de Oliveira, A.G. Prevalence of silent vertebral fractures detected by vertebral fracture assessment in young Portuguese men with hyperthyroidism. Eur. J. Endocrinol. 2015, 172, 189–194. [Google Scholar] [CrossRef]
- Vestergaard, P.; Mosekilde, L. Fractures in patients with hyperthyroidism and hypothyroidism: A nationwide follow-up study in 16,249 patients. Thyroid 2002, 12, 411–419. [Google Scholar] [CrossRef]
- Vestergaard, P.; Rejnmark, L.; Mosekilde, L. Influence of hyper- and hypothyroidism, and the effects of treatment with antithyroid drugs and levothyroxine on fracture risk. Calcif. Tissue Int. 2005, 77, 139–144. [Google Scholar] [CrossRef]
- Dhanwal, D.K.; Gupta, N. Bone mineral density trends in Indian patients with hyperthyroidism—Effect of antithyroid therapy. J. Assoc. Physicians India 2011, 59, 561–567. [Google Scholar]
- Deshmukh, H.; Papageorgiou, M.; Aye, M.; England, J.; Abdalla, M.; Sathyapalan, T. Hyperthyroidism and bone mineral density: Dissecting the causal association with Mendelian randomization analysis. Clin. Endocrinol. 2021, 94, 119–127. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mudri, D.; Bilić Ćurčić, I.; Meštrović, L.; Mihaljević, I.; Kizivat, T. Hyperthyroidism and Wnt Signaling Pathway: Influence on Bone Remodeling. Metabolites 2023, 13, 241. https://doi.org/10.3390/metabo13020241
Mudri D, Bilić Ćurčić I, Meštrović L, Mihaljević I, Kizivat T. Hyperthyroidism and Wnt Signaling Pathway: Influence on Bone Remodeling. Metabolites. 2023; 13(2):241. https://doi.org/10.3390/metabo13020241
Chicago/Turabian StyleMudri, Dunja, Ines Bilić Ćurčić, Lucija Meštrović, Ivica Mihaljević, and Tomislav Kizivat. 2023. "Hyperthyroidism and Wnt Signaling Pathway: Influence on Bone Remodeling" Metabolites 13, no. 2: 241. https://doi.org/10.3390/metabo13020241
APA StyleMudri, D., Bilić Ćurčić, I., Meštrović, L., Mihaljević, I., & Kizivat, T. (2023). Hyperthyroidism and Wnt Signaling Pathway: Influence on Bone Remodeling. Metabolites, 13(2), 241. https://doi.org/10.3390/metabo13020241