Abstract
Prostate cancer (PCa) is the common cause of death in men. The pathophysiological factors contributing to PCa are not well known. PCa cells gain a protective mechanism via abnormal lipid signaling and metabolism. PCa cells modify their metabolism in response to an excessive intake of nutrients to facilitate advancement. Metabolic syndrome (MetS) is inextricably linked to the carcinogenic progression of PCa, which heightens the severity of the disease. It is hypothesized that changes in the metabolism of the mitochondria contribute to the onset of PCa. The studies of particular alterations in the progress of PCa are best accomplished by examining the metabolome of prostate tissue. Due to the inconsistent findings written initially, additional epidemiological research is required to identify whether or not MetS is an aspect of PCa. There is a correlation between several risk factors and the progression of PCa, one of which is MetS. The metabolic symbiosis between PCa cells and the tumor milieu and how this type of crosstalk may aid in the development of PCa is portrayed in this work. This review focuses on in-depth analysis and evaluation of the metabolic changes that occur within PCa, and also aims to assess the effect of metabolic abnormalities on the aggressiveness status and metabolism of PCa.
1. Introduction
There is no universal definition for cancer, but it is defined as the uncontrol division of the cell that leads to invading the cell basement or metastasizing to other body organs [1,2,3]. Benign and malignant tumors can be differentiated based on their location. A benign tumor remains at the primary site. In contrast, malignant tumor cells invade the cell membrane and enter the blood vessel via intravasation [2].
The benign prostate tumor might limit glycolysis and favor high oxidative phosphorylation. In contrast, the increased glycolysis becomes an advanced castrate resistance PCa characteristic. Changes occur in prostate epithelial cells—a unique metabolic phenotype, during the movement of the tumor from prostatic intraepithelial neoplasia (PIN) to metastasis. In that case, normal prostate epithelial cells depend on glycolysis and use glucose to produce citrate. Simultaneously, PCa cells gradually re-activate mitochondrial phosphorylation with glucose metabolism, reducing citrate production. Androgen receptor (AR) in PCa supports metabolic and biosynthetic demands by reprogramming cellular metabolic pathways: mitochondrial respiration, aerobic glycolysis, and de novo lipogenesis [4,5,6,7]. Fatty acid (FA) synthesis may be an early event in prostate tumor formation and is also linked with disease progression [8,9]. Amino acid (AA) metabolism maintains the AA pool required in PCa progression. Glucose, lipids, and nitrogen precursor are purines and pyrimidines required for nucleic acid (NA) synthesis. AA synthesis and its metabolism in PCa focused on anaplerosis compared to energy production [10,11].
Several studies have focused on the characterization of PCa cell metabolic profile and pointed to the biological mechanism involved in disease progression. A study by Gómez-Cebrián et al., 2020, reported that healthy prostate cells show reduced citrate oxidation with lowering m-aconitase (ACO) activity due to the effect of high zinc concentration. It also slower TCA cycle metabolism [12]. Healthy prostate cells mostly rely on glycolytic pathways for energy [13,14]. The ultimate read-out of gene-environment interactions is obtained by measuring many low molecular weight substances (metabolites) in biofluids using metabolomics which may help identify novel risk factors for PCa. Several studies investigated the relationship between pre-diagnostic plasma, serum metabolite level, and PCa risk incidence [15,16,17,18].
2. A Link between PCa and MetS
MetS-like insulin resistance (IR), adipose tissue generated adipocytokines, and obesity are the factors that play a significant role in cancer development [19]. According to recent studies by Gacci et al. and Avgerinos et al., the mechanism and metabolic pathways are not fully characterized [20,21]. However, MetS and Cancer have some associations, including alteration in IGF-1 synthesis and signaling pathways due to IR [20], overproduction of sexual hormones [21,22], adipose tissue accumulation [20], fluctuations in sleep patterns, and abrupt dietary changes [21]. This factor leads to cancer cell proliferation and survival by alterations in an increase in certain events, such as proliferation and decreased apoptosis. This evidence is a considerable link between obesity and cancer caused by chronic inflammation, microbiome dysregulation, and IR [20]. MetS appear as a significant influencer in cancer cell progression. For example, PCa with MetS appears to increase cell death chances with high-grade tumor amplification [21].
MetS could be the potential risk factor for PCa development. However, previous studies reported an inconsistent link between PCa and MetS [23]. Simultaneously, some studies reported that MetS could increase PCa development [24,25]. In contrast, some studies reported no relation and negative association between MetS and PCa [26]. Recently, a study by Hammarsten et al. reported that MetS and its components might reduce the serum prostate-specific antigen (PSA) [27]. Gao et al. conducted the study in a Chinese cohort to explore the relationship between MetS and PCa. They found that the PCa incidence in men over 40 years of age was 0.1%. Based on this, it was concluded that MetS and obesity were not the PCa risk factor, whereas PSA levels with high age were the risk factor for PCa [28]. This relationship is controversial because, in Asian population studies, these inconsistencies have also been observed [29].
3. MetS and Incidence of PCa
PCa is the third most common malignancy and the second leading cause of death in the US [30]. According to data from 2020, it is estimated that there are 1.4 million diagnosed cases with an average age of 66 years and 360 thousand death/year with PCa. Additionally, countries such as the Caribbean, Australia, Europe, North America, and South Africa have a positive and negative relationship with PCa with high frequency [31,32,33,34]. It grows in an androgen-dependent state, and survival is based on androgen deprivation therapy (ADT)—the first line of treatment for advanced disease [35]. Studies also reported that MetS is linked with a high risk of PCa development and progression [36,37].
An international study has confirmed that in 32 out of 40 countries, the incidence of PCa is increasing, while in another eight countries is relatively stable [38,39]. According to a recent meta-analysis, MetS significantly increased the high-grade PCa incidence while having little impact on the prevalence of PCa [21]. Additionally, another study revealed that racial and geographic factors influence the association between MetS and PCa. MetS raises the risk of PCa in European countries, whereas it has little to no impact on PCa risk in the United States and other Asian countries [40]. A prospective population-based study conducted in Finland with 1880 males who had no prior history of diabetes or cancer at baseline and were followed for an average of 13.2 years stated that the individual factor of the MetS has been related to an elevated risk of PCa [41]. In a clinical trial, the patients with no prior history of PCa were screened to identify the link between PCa risk and MetS via transrectal ultrasound-guided prostate biopsy [42]. Lifestyle changes may alter the MetS-related disease progression. This may also treat or prevent the disease condition [43,44] (Table 1).

Table 1.
MetS and PCa studies. This table summarizes epidemiological studies carried out on MetS and PCa relations.
4. MetS-Like Components on PCa Development
Obesity, diabetes, hypertension, and hypercholesterolemia are the MetS-like components [52]. Whereas variables such as elevated fasting glucose, triglycerides, and high blood pressure are not identical to the classic definition of MetS [53]. Hypertension and abdominal obesity are common in PCa patients, whereas diabetes is not linked with PCa risk. Beebe-Dimmer et al. reported that PCa-associated MetS in American-African men differs by race [24]. The study by Sourbeer et al. stated that more than three MetS-components are associated with high-grade PCa, not with an overall or low-grade PCa [53,54]. However, several studies about the relationship between MetS and PCa have been inconsistent [23]. Additional epidemiologic data are needed if Mets is a risk factor for PCa development [26]. Future studies must determine whether to prevent MetS and reduce the risk of aggressive PCa.
Several biological mechanisms explain that MetS can lead to an increased risk of high-grade PCa, e.g., high cholesterol levels [55]. The altered level of IGF-1 [56], leptin [57], and adiponectin [58] in MetS conditions can also be linked with PCa risk. The pro-inflammatory state, including the increased C-reactive protein (CRP) level, interleukins-18 (IL-18), IL-1β, IL-6, and Tumor necrosis factor-α (TNF-α), is associated with MetS, which has been ultimately linked to PCa [59,60,61]. A MetS-linked component type II diabetes is related to low PCa risk due to pancreatic β-cells damage mediated hypoinsulinemia [62,63]. In contrast, hyperinsulinemia (HI) is associated with a high risk of PCa death [64]. These are the conditions making a clear association of MetS with PCa. There is a need for more extensive studies with multiethnic groups.
5. The Metabolic Phenotype of PCa
Metabolic phenotyping has developed into an effective method for discovering novel molecular biomarkers and metabolic vulnerabilities that may represent novel therapeutic options in cancer diseases [65,66]. The zinc (Zn) deposition in prostate cells leads to mitochondrial aconitase (ACO2) inhibition, lowering the citrate oxidation and consequently decreasing tricarboxylic acid (TCA) cycle metabolism [10]. In response to low Zn levels in PCa, reverse this condition [13]. With these conditions, it is reported that TCA cycle metabolism in PCa leads to decreased citrate levels and increased TCA cycle intermediates, including malate, fumarate, succinate ect [67,68,69]. The monocarboxylate transporters (MCT) expression levels differ PCa patients. MCT-producing phenotype related to aggressive PCa. Inhibiting its activity leads to a faster accumulation of toxic metabolic products [70]. The lactate shuttle expression in PCa can be a potential biomarker for the diagnosis and prognosis of the disease [71]. Glutaminase-1 performs a glutaminolysis, while its expression is upregulated in PCa. Blocking its action may dysregulate glutamine-based energy production in PCa [72,73]. Studies have shown that arginine maintains the malignant phenotype in PCa and is also needed for PCa growth, while the exact mechanism is not well understood [74]. PCa cells generate fatty acids by de novo lipid synthesis to obtain energy. This change to a lipid-producing phenotype marks a significant turning point in the development of PCa [13,75]. Warburg effect is characterized by alteration in preferential energy-producing pathways. The cancer cells follow aerobic glycolysis to produce ATP, while normal cells follow oxidative phosphorylation for ATP production [76,77].
6. Metabolic Regulation of PCa
The mechanisms involved in the transformation stages of PCa are not well understood. Metabolic reprogramming of cancer cells characterizes malignant changes, which can also become increasingly apparent. The molecular aspects of this reprogramming vary between cancer types and individual cells within a malignancy. Using glucose to generate citrate that is released as part of the seminal fluids is an unusual and inefficient energy metabolism for normal prostate epithelial cells. During transformation, PCa cells switch from an inefficient to efficient energy metabolism [3,78,79,80,81] (Figure 1).
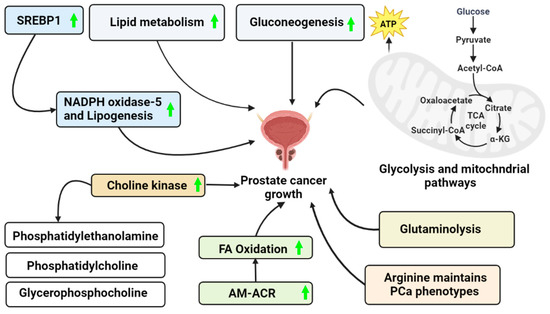
Figure 1.
PCa growth via a different mechanism. This figure represents the possible ways to acquire the energy required to maintain PCa cell survival in an ATP-depleted cell environment. (Green arrow—Upregulation).
6.1. Glycolysis
Positive [vascular endothelial growth factor [VEGF], IL-8, and stromal cell-derived factor [SDF]-1/(CXCL12)] and negative regulation of the angiogenic cascade are examples of hypoxia-inducible genes that are known to play an essential role in the initial stages of tumor adaptation to hypoxia. Hypoxia-inducible genes similarly regulate necessary glycolytic enzymes for anaerobic metabolism. These molecules are essential for producing growth-related macromolecules and fulfilling the high-energy needs of expanding tumors [82]. As a result, the so-called “Warburg effect,” in which cancer cells switch from producing energy via oxidative phosphorylation to doing so via glycolysis, is widely believed to be an essential feature of cancer cells and is linked to more rapid tumor growth [83,84,85].
One of the enzymes in the glycolytic process, phosphoglycerate kinase (PGK1), generates ATP. In PCa, PGK1 is frequently overexpressed and is controlled by hypoxia-inducible factor-1a (HIF-1a) [86,87]. PGK1 maps to a region of the X chromosome (Xq11–Xq13) [88] that has been linked to an increased risk of developing PCa, hypospadias, and androgen insensitivity in families. PGK1 plays a role in DNA replication and repair by acting on DNA in the nucleus. Surprisingly, tumors extracellularly release PGK1, which can act as a disulfide reductase. It has the potential activity to cleave plasminogen to yield angiostatin (a vascular inhibitor) [89,90,91,92,93,94]. Therefore, it is probable that overexpression of PGK1 will slow tumor growth by reducing angiogenesis. Due to this, tumor growth may depend on a finely tuned balancing act between the hypoxic response, which is required to produce pro-angiogenic factors and events necessary for anaerobic metabolism (such as PGK1). Controlling PGK1 secretion is a significant obstacle. According to recent investigations, adipocytes, which have been demonstrated to control metabolism in the primary tumor via CAFS, are possibly responsible for the transition to the Warburg effect in PCa metastatic cells. High glycolytic rates and increased HIF1 synthesis result from PCa cells co-culturing with adipocytes without oxygen. The accumulation of lactate and the inhibition of oxidative phosphorylation (OXPHOS) are caused by the transcription of the Warburg-associated genes driven by the HIF1 increase. However, PCa cells treated with CM from PCa/adipocyte co-culture undergo remarkable metabolic reprogramming. However, PCa CM-treated adipocytes release a substantial amount of free glycerol and show an increase in the lipolytic enzyme adipose triglyceride lipase (ATGL), indicating a PCa-induced lipolytic phenotype [7,95]. Despite the lack of experimental proof in this scenario, it is well-established that glycerol participates in the glycolytic process to provide energy for cancer cells [96,97,98]. Altogether, established results support the hypothesis that PCa cells tamper with their surroundings to produce metabolic intermediates utilized by cancer cells. Consequently, the tumor’s microenvironment provides metabolic support for the growth of the tumor, making it an optimal setting for the survival and proliferation of cancer cells [78,99].
Citrate can meet PCa cancer cells’ energy needs because citrate excretion is downregulated [100]. Schöpf et al., 2020 [101] found that glutamate and malate drove OXPHOS capacity in benign human prostate tissue, while in malignant tissue, succinate and pyruvate drove energy production and compensated for the diminished N-pathway capacity, correlating with results from PCa cell lines by Weber et al., 2018 [102]. Researchers Badder et al. (2019) [103] found that OXPHOS activity in PCa cell models revealed the significance of pyruvate in maintaining tumor growth. Recent in vitro research by Zadra et al., 2015 shows that modifying AMPK’s metabolic activity slows the development of PCa [104]. Inhibiting glutamine absorption reduces PCa cell proliferation and invasion, according to an in vitro study by Wang et al., 2015 [73].
6.2. Gluconeogenesis
It is generally known that many cells can create energy (ATP) from alternate fuels, such as ketone bodies or fatty acids, in functioning mitochondria, even when glucose is scarce. [105,106]. Since the gluconeogenic and glycolytic processes exchange intermediates, gluconeogenesis could be a backup source of biosynthetic precursors when glucose is scarce [107,108].
The availability of nutrients is a constant source of stress for cancer cells, and disrupting their adaptive responses could be a proper treatment strategy. Cancer cells have been found to generate critical metabolites using shortened gluconeogenesis versions by expressing phosphoenolpyruvate carboxykinase (PEPCK, PCK1, or PCK2). Gluconeogenesis is the metabolic route that branches off glycolysis and employs lactate or amino acids as substrates. There is evidence that PCK1 and PCK2 are essential for developing several malignancies. In contrast, the downstream gluconeogenesis enzyme fructose-1,6-bisphosphatase 1 (FBP1) suppresses glycolysis and tumor growth through non-enzymatic processes. It has been found that PCK2 expression in PCa metastases is higher than in normal prostate cells or primary tumors [109]. Overall survival was significantly lower in PCa patients with highly expressed PCK2. In this work, PCa tumor-initiating cells (TIC) expressed considerably more PCK2 than other PCa cell types. Silencing PCK2 decreased the number of TIC, reduced their sphere-forming ability, and stopped the formation of PCa nodules in mice. Silencing PCK2 decreased glycolysis, indicating that PCK2 plays a role in driving the switch to glycolysis. Notably, TIC survival was marginally improved by PCK2 silencing in a glucose-free media. Acetyl-CoA levels, measured by the acetylation of lysine residues in histones and total proteins, were increased after PCK2 silencing. It was owing to increased synthesis from citrate via ATP citrate lyase (ACLY) [109,110].
6.3. Lipid Metabolism in PCa
Researchers have investigated the involvement of lipid metabolism in the development and progression of PCa [111]. It has a crucial role in the malignant phenotype expression [112,113]. During PCa development, PCa cells undergo adaptive metabolic alterations to maintain growth and progression [114]. The high expression and activity of cell membrane phospholipids biosynthesis enzyme known as choline kinase with other lipid molecules, including phosphatidylethanolamine, phosphatidylcholine, and glycerophosphocholine, are reported in PCa [115]. The α-methyl acyl-CoA racemase (AM-ACR) is the peroxisomal enzyme required to oxidize branch-chained fatty acids (FAs). AM-ACR is highly expressed in PCa cells and increases FAs oxidation as an energy source to support PCa progression and survival [116,117,118].
Huang et al. stated that sterol regulatory binding protein 1 (SREBP-1) increases the NADPH oxidase-5 and lipogenesis, which plays a significant role in PCa growth, and metastasis [119]. The lipogenic phenotype of PCa is associated with de novo lipogenesis, and cholesterogenesis is carried out by converting derived citrate from the TCA cycle and oxaloacetate to acetyl-Co-A by ACLY in the cytosol. Acetyl-CoA carboxylase (ACC) converts acetyl-CoA to malonyl-CoA and oxaloacetate into pyruvate, which can re-enter the mitochondria for further utilization. This leads to high activation of de novo lipogenesis and cholesterogenesis. Whereas it has been found that there is increased expression of ACC, ACLY, and FA synthase in PCa [120,121].
6.3.1. Cholesterol Metabolism
The plasma membrane contains cholesterol, approximately one-third of lipids [122]. Cholesterol is crucial for cell proliferation, while its depletion leads to cell arrest [123]. It maintains cancer stem cells and activates oncogenic Hedgehog signaling [124,125]. Compared with normal cells, cholesterol is highly needed for PCa cell proliferation [126,127]. The elevated cholesterol level was transferred to high-density lipoprotein (HDL) particles by ATP-binding cassette transporter A1 (ABCA1) and G1 (ABCG1) [128]. Whereas the excessive intracellular cholesterol returns to the intestine or liver by HDL particles, where it is recycled. It is delivered to steroidogenic organs for hormone synthesis [129,130]. Acyl-Co acyltransferases esterify excessive free cholesterol and are stored in cytoplasmic lipid droplets to avoid cholesterol toxicity. They are exported from cells via ABCA1 and ABCG1 [128,131]. PCa can be easily diagnosed with high cholesterol levels due to elevated PSA. However, the studies on these did not consider [132]. Whereas highly expressed LDLR in PCa cannot uptake enough cholesterol from the blood. Therefore, the cholesterol level in the blood is the same before and after prostate surgery [133,134,135].
Cholesterol metabolism is associated with PCa pathogenesis and acts as a substrate for intratumoral androgen biosynthesis [136]. Castration-resistant PCa cells (CRPCa) are highly expressed CPY17A1 enzymes that de-novo synthesizes androgens [137]. It emphasizes the significance of steroid biosynthesis as a critical biological process connecting PCa and cholesterol [115,138]. The high-grade PCa with serum cholesterol level gives a positive correlation [139], several cholesterol metabolic regulator abnormalities found in in-vivo [140] and in-vitro cancer progression models [141,142], and statin lowering the risk of PCa occurrence, which also be used for a cholesterol-lowering therapy [143,144,145].
6.3.2. Caveolin-1-Mediated Metabolism
Caveolae formation requires Polymerase-I and transcript release factor (PTRF) as CAV1 and CAVIN1. These are the isoform of Cav-1 found in Cav-α and β. Studies identified Cav-1 as a substrate for proto-oncogenic kinases ABL1 [146], FYN [147], and SRC [148]. It can be phosphorylated in response to rapamycin (mTOR) complex 2 (mTORC2) signaling [149] and epithelial growth factor (EGF) [150]. Caveolin-1 (Cav-1) is also known as a lipid chaperone. It is a transporter molecule that facilitates the mechanoprotection of cell membranes, cellular lipid homeostasis, and endocytosis and exocytosis [151]. Similarly, it modulates transmembrane signal transduction and microdomain arrangement in the membrane [152]. It plays a role in transporting chemokines, proteins, LDL, and HDL [153].
Cav-1 disturbs several metabolic pathways in PCa cells [154]. It has a dynamic role in cancer [155]. It is found in the activation and regulation of integrins and cadherins, receptor tyrosine kinases (RTK). G-protein coupled receptors (GPCR) [156]. High expression of Cav-1 is linked with aggressive tumor phenotypes [157] and metastasis potential [158]. It is also associated with radio drug and multidrug resistance [159]. Tumor-supportive oncometabolism studies stated that Cav-1 enables re-modification of cancer cell lipid metabolism toward increased sphingomyelins catabolism to ceramide derivatives and changes ceramide metabolism in cancer cells. These all lead to the efflux of Cav-1-sphingolipid particles containing mitochondrial proteins and lipids and increased glycosphingolipid synthesis [160].
6.4. Mitochondrial Metabolism in PCa
Mitochondrial is also known as “the powerhouse of the cell” and plays a major role in reprogramming cancer cell metabolism [161]. It is an important bioenergetic hub that maintains ATP production, ROS generation, calcium signaling, redox balance, and apoptotic pathways [162,163]. Almost all metabolic fuels combine with acetyl-Co-A and can be converted entirely into CO2, water, and ATP, directing it into catabolic processes. The major metabolic pathways in mitochondria involve the catabolism of biomolecules and energy production, such as the TCA cycle, the electron transport chain (ETC), FAO, and OXPHOS [164].
In PCa metabolism, glucose and aspartate produce citrate and are secreted in prostatic fluid. The normal prostate epithelial cells use glucose to maintain their physiological citrate secretion, which is oxidized in the TCA cycle in PCa. In contrast, pyruvate is the primary source in the TCA cycle of PCa cells. During the PCa cell transformations, PCa cells consumed citrate to enhance OXPHOS and used it to fuel lipogenesis [6]. Understanding how each of these mechanisms works in various cancer types may offer novel therapy ideas as they all have potential roles in the development and malignant phenotypes maintenance [165] (Figure 2).
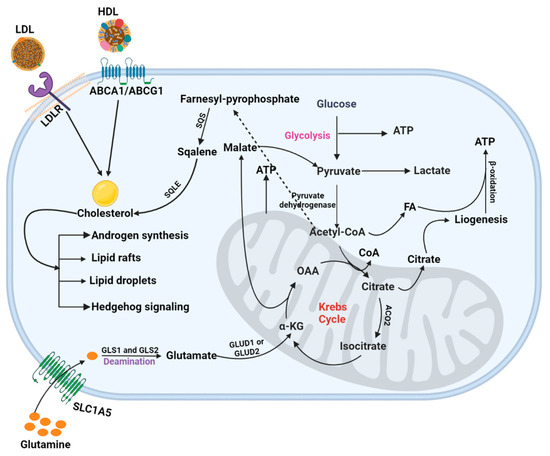
Figure 2.
PCa metabolism. This figure illustrated the mechanistic pathways, including glycolysis, TCA cycle, and cholesterol and glutamine metabolism, involved in energy production in PCa cells.
Multiple investigations have shown that NF-κB aids PCa development and progression by encouraging cell survival, proliferation, and invasion [166,167,168]. PCa has been linked to multiple members of the NF-κB family, although p52 is particularly crucial. RelA, RelB, and c-Rel are all transactivating subunits [169,170,171,172,173]. In particular, Bcl-3 is required for the survival of PCa cells after chemotherapy [174]. It has been hypothesized that so-called cancer stem cells are essential for the emergence of resistance to treatment, and a population of AR-negative PCa stem cells with constitutive NF-κB activity was lately identified [175,176]. Castration or AR antagonist resistance is primarily attributable to NF-κB, and promising outcomes have been shown in combination treatments that target both NF-κB and AR antagonists [177,178,179]. Aspirin and its active metabolite salicylic acid (SA) has been shown to effectively suppress the growth of the androgen-independent PCa cell line DU-145, adding further evidence for the significance of NF-κB in PCa proliferation [180]. However, aspirin may affect PCa by inhibiting cyclooxygenase (COX), the enzyme responsible for prostaglandin formation. Prostaglandins are crucial for the proliferation of PCa cells [181,182,183]. SA’s direct effect on NF-κB signaling at several levels is the most likely explanation for free SA’s inhibitory actions, as free SA cannot inhibit COX [184]. High mobility group box protein 1 (HMGB1) overexpression is correlated with tumor cell proliferation and aggressiveness in PCa [185,186,187]. Long-term Aspirin or NSAIDS use has been shown to reduce the risk of PCa in epidemiological studies [187]. AR inhibits expression induced by canonical NF-κB (RelA/p50) but appears to increase activation of non-canonical NF-κB favorably [188]. Loss of androgen repression of NF-κB target genes is related to poor prognosis in metastatic PCa [189], suggesting that activation of non-canonical NF-κB may be a crucial stage in establishing androgen independence. In addition to inducing metabolic reprogramming of PCa cells via activation of genes for glucose absorption and metabolism [169,190], the p52 subunit can also stimulate AR signaling, which adds to androgen-independent proliferation [191,192]. But there may be yet another layer of crosstalk between NF-κB and AR, as the classical IKKα and IKKβ upstream of NF-κB may also effectively regulate AR activation through phosphorylation [193,194,195,196,197,198].
Glutamine Metabolism
Glutamine (Gln) is synthesized by the human body. It is an essential source for normal and cancer cell survival, as they will die under Gln-depletion conditions [199,200,201]. Under the catabolic condition, it is consumed by the gastrointestinal (GI) tract, kidney, and immune compartment. It is an energy source for biosynthesis and homeostasis [202,203,204]. An in-depth understanding is required for regulating Gln-metabolism in cancer cell development and proliferation associated with hormonal studies such as progesterone, thyroid hormone, androgen, estrogen, prostaglandin, and insulin [205].
Previously, Gln was found to maintain the redox state by reducing ROS generation and producing GSH in PCa [199,206]. Lipids, pyruvate, and succinate appear as the primary energy source and biosynthesis in PCa. Gln has less impact in low-risk primary PCa [136,137] and has a prominent role in advanced PCa [207]. The PCa with high MYC expression acquired oncogene-driven Gln addiction due to the high demand for Gln for cancer cell progression [200,208]. It plays a significant role in ATP production by participating in the TCA cycle [72,204,206]. Gln enters the mammalian cells through AA transporters such as SLC1A5/ASCT2 and undergoes deamination in the mitochondrial cell by the action of glutaminase-1 (GLS-1) (kidney type glutaminase) and GLS-2 (liver type glutaminase), and finally transformed into glutamate (Glu) [72,209]. Next, by the action of glutamate dehydrogenase (GLUD1 or GLUD2) on Glu, it is converted into α-ketoglutarate (α-KG). It participates in ATP, NADH, and FADH2 production by entering the Kreb cycle. Additionally, malate (a Kreb cycle intermediate) leaves the process. It produces pyruvate and NADPH [210], whereas oxaloacetate (OAA) is used for nucleotide synthesis by converting it to aspartate. Citrate undergoes cataplerosis and the production of acetyl-CoA and lipids [202,211]. Therefore, Gln-metabolism benefits PCa by enhancing tumor development.
6.5. Neuroendocrine PCa (NEPCa) Metabolism
NEPCa is a newly arisen, more often form of aggressive disease. Androgen deprivation leads to reduce AR signaling in NEPCa. The NEPCa are also distinguished for their high neuroendocrine lineage markers expression, which are synaptophysin, chromogranin-A, and enolase [10,212]. Compared to prostate adenocarcinoma, mutational genetic changes in Rb1 and Tp53 and MYCN and AURKA amplification are more dominant in NEPCa. At the same time, MYCN is involved in neuroendocrine lineage reprogramming. It increases histone acetylation with mitochondrial export of acetyl groups and DNA accessibility [213]. Functional loss of Rb1 and Tp53 enabled the pluripotency networks activations via expressional activation of SOX2 (a transcriptional factor), epigenetic modifier, and enhancer of zeste-homolog 2 (EZH2) [214,215]. In glycolysis, pyruvate is generated and converted to acetyl-CoA, which plays a central role in regulating histone acetyltransferase (HAT) enzyme activity. In treatment, the high expression of histone lysine demethylase KDM8 induces NEPCa tumor to reprogram metabolism towards aerobic glycolysis [216,217]. The enhanced glutamine uptake and increased glycolysis elevate the pyruvate and acetyl-CoA production in NEPCa [218]. The highly activated glycolysis associated with MCT-4 mediates lactic acid regulation. It is the most clinically relevant metabolic feature in NEPC [219].
7. Effect of Myokines in PCa
Myokines are the cytokines produced by skeletal muscle and travel through a circulatory system, such as growth factors, metallopeptidases, and several other factors [220,221,222]. These factors may have beneficiary effects on the liver, reducing insulin resistance (IR) and adiposity, the immune system, and improving glucose uptake [223]. Their levels keep on changing as IL-6 [224,225], IL-10, IL-15 [226], oncostatin M [227] and decorin [228], irisin [229], myostatin, and secreted protein acidic risk in cysteine (SPARC) [230,231] may suppressing cancer cell proliferation and inhibiting the epithermal to mesenchymal cells transition (EMT), also accelerating apoptosis via cell cycle arrest. During exercise, skeletal muscle produces secret fibroblast growth factor-21 (FGF21), which can protect against low-grade systemic inflammation and reduce T2D risk via upregulating glycogenesis, downregulating gluconeogenesis, and lipogenesis in the liver [223]. IR, HI, and hyperlipidemia are associated with obesity, generating a tumor-favorable environment (TFE). Exercise-induced myokines can influence TFE by controlling adipose tissue and adipocytes [232]. It suggests that myokines have a potential protective effect on cancer. However, limited evidence suggests a direct association with tumor suppression [231,233,234].
8. Role of Androgens in PCa
In normal prostate cells, Zn accumulation inhibits m-aconitase, which blocks the citrate to isocitrate conversion in the TCA cycle. Androgen carries out this process and results in a disturbed TCA cycle with an elevated citrate level in prostatic fluid. Whereas Zn concentration is low in the PCa and cannot be accumulated for a long time. Due to this, the m-aconitase stays free and can regulate the TCA cycle to derive the energy and acetyl-CoA (anabolic substrate) for de novo lipogenesis [37,100].
Androgen drives the PCa cell proliferation and survival via the AR axis. It regulates cellular metabolism, including homeostasis, tissue differentiation, and lipid metabolism [235]. In addition, promoting expression and activation of the transcription factor is sterol regulatory element-binding proteins (SREBPs). SREBPs bind to fatty acids (FAs) in the gene promoter regions [236]. Androgen also has direct AR-binding sites in the promoter region of the FA synthase gene, and it may extend to many other lipogenic enzymes [237,238]. Androgens stimulate lipid metabolism enzymes’ expression and influence lipid profiles in PCa cells [120,239]. Researchers worked on elucidating androgen-mediated FA uptake and oxidation with modulating FA transporters in PCa proliferation [240,241]. De novo lipogenesis might aid PCa growth by supplying the raw materials needed for synthesizing membranes and signaling. PCa cells may prefer FAO to support their viability after tumors expand, their vascular network compromising access to oxygen and nutrients. Following dispersion, PCa cells may adapt their metabolism to favor aerobic glycolysis. At the same time, FAO and de novo lipogenesis remain highly active [95,242].
9. Conclusions
Cancer is the most common cause of death worldwide, so it is critical to develop innovative solutions to this issue. Humanity would have sensitive tools for detecting and effectively treating it in an ideal world. Therefore, fast and accurate diagnostic techniques are required for its detection [243,244,245].
We are learning more and more about the complex molecular mechanisms that underlie PCa metabolism. Characterizing and regulating PCa’s metabolism may be crucial for delivering individualized treatment for the disease. Preventing risk factors might be a valuable strategy in the urgent demand for novel medicines. MetS seem to have the opposite impact. Therefore, more research is required to determine the relationship between MetS and the development of PCa. Several metabolic changes occur during the development of a benign cell into a malignant cell. Several cellular cancer metabolism mechanisms are still poorly known, requiring a lot of research. Better diagnostic procedures and treatment options may be developed due to increased understanding.
Author Contributions
Conceptualization, U.R.W., A.G.M., A.V.G. and R.M.; resources and data curation, R.M., A.D. and B.V.; writing—original draft preparation, U.R.W., A.G.M. and R.M.; writing—review and editing, U.R.W., A.G.M., A.V.G. and R.G.; supervision, A.V.G. and R.G.; project administration, A.V.G. and R.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Data are available from the authors on request (A.V.G.) due to privacy or ethical restrictions.
Acknowledgments
The authors thank the VIT, Vellore, Tamilnadu, India, for supporting this work.
Conflicts of Interest
The authors declared no potential conflict of interest concerning this article search, authorship, and publication.
References
- Hausman, D.M. What Is Cancer? Perspect. Biol. Med. 2019, 62, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Lei, K.F.; Han, F. Tumor microenvironment: Recent advances in various cancer treatments. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3855–3864. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.G.; Wanjari, U.R.; Namachivayam, A.; Murali, R.; Prabakaran, D.S.; Ganesan, R.; Renu, K.; Dey, A.; Vellingiri, B.; Ramanathan, G.; et al. Role of Immune Cells and Receptors in Cancer Treatment: An Immunotherapeutic Approach. Vaccines 2022, 10, 1493. [Google Scholar] [CrossRef]
- Audet-Walsh, É.; Dufour, C.R.; Yee, T.; Zouanat, F.Z.; Yan, M.; Kalloghlian, G.; Vernier, M.; Caron, M.; Bourque, G.; Scarlata, E.; et al. Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer. Genes Dev. 2017, 31, 1228–1242. [Google Scholar] [CrossRef]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef]
- Mamouni, K.; Kallifatidis, G.; Lokeshwar, B.L. Targeting Mitochondrial Metabolism in Prostate Cancer with Triterpenoids. Int. J. Mol. Sci. 2021, 22, 2466. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.G.; Wanjari, U.R.; Prabakaran, D.S.; Ganesan, R.; Renu, K.; Dey, A.; Vellingiri, B.; Kandasamy, S.; Ramesh, T.; Gopalakrishnan, A.V. The Cellular and Molecular Immunotherapy in Prostate Cancer. Vaccines 2022, 10, 1370. [Google Scholar] [CrossRef]
- Flavin, R.; Zadra, G.; Loda, M. Metabolic alterations and targeted therapies in prostate cancer. J. Pathol. 2011, 223, 283–294. [Google Scholar] [CrossRef]
- Czernin, J.; Benz, M.R.; Allen-Auerbach, M.S. PET Imaging of Prostate Cancer Using C-Acetate. PET Clin. 2009, 4, 163–172. [Google Scholar] [CrossRef]
- Ahmad, F.; Cherukuri, M.K.; Choyke, P.L. Metabolic reprogramming in prostate cancer. Br. J. Cancer 2021, 125, 1185–1196. [Google Scholar] [CrossRef]
- Strmiska, V.; Michalek, P.; Eckschlager, T.; Stiborova, M.; Adam, V.; Krizkova, S.; Heger, Z. Prostate cancer-specific hallmarks of amino acids metabolism: Towards a paradigm of precision medicine. Biochim. Biophys. Acta. Rev. Cancer 2019, 1871, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Cebrián, N.; García-Flores, M.; Rubio-Briones, J.; López-Guerrero, J.A.; Pineda-Lucena, A.; Puchades-Carrasco, L. Targeted Metabolomics Analyses Reveal Specific Metabolic Alterations in High-Grade Prostate Cancer Patients. J. Proteome Res. 2020, 19, 4082–4092. [Google Scholar] [CrossRef]
- Eidelman, E.; Twum-Ampofo, J.; Ansari, J.; Siddiqui, M.M. The Metabolic Phenotype of Prostate Cancer. Front. Oncol. 2017, 7, 131. [Google Scholar] [CrossRef]
- Lima, A.R.; Araújo, A.M.; Pinto, J.; Jerónimo, C.; Henrique, R.; Bastos, M.L.; Carvalho, M.; Guedes de Pinho, P. GC-MS-Based Endometabolome Analysis Differentiates Prostate Cancer from Normal Prostate Cells. Metabolites 2018, 8, 23. [Google Scholar] [CrossRef]
- Röhnisch, H.E.; Kyrø, C.; Olsen, A.; Thysell, E.; Hallmans, G.; Moazzami, A.A. Identification of metabolites associated with prostate cancer risk: A nested case-control study with long follow-up in the Northern Sweden Health and Disease Study. BMC Med. 2020, 18, 187. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Mondul, A.M.; Weinstein, S.J.; Koutros, S.; Derkach, A.; Karoly, E.; Sampson, J.N.; Moore, S.C.; Berndt, S.I.; Albanes, D. Serum metabolomic profiling of prostate cancer risk in the prostate, lung, colorectal, and ovarian cancer screening trial. Br. J. Cancer 2016, 115, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.A.; Fensom, G.K.; Rinaldi, S.; Scalbert, A.; Appleby, P.N.; Achaintre, D.; Gicquiau, A.; Gunter, M.J.; Ferrari, P.; Kaaks, R.; et al. Pre-diagnostic metabolite concentrations and prostate cancer risk in 1077 cases and 1077 matched controls in the European Prospective Investigation into Cancer and Nutrition. BMC Med. 2017, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.A.; Fensom, G.K.; Rinaldi, S.; Scalbert, A.; Appleby, P.N.; Achaintre, D.; Gicquiau, A.; Gunter, M.J.; Ferrari, P.; Kaaks, R.; et al. Patterns in metabolite profile are associated with risk of more aggressive prostate cancer: A prospective study of 3057 matched case-control sets from EPIC. Int. J. Cancer 2020, 146, 720–730. [Google Scholar] [CrossRef]
- Torres, S.; Fabersani, E.; Marquez, A.; Gauffin-Cano, P. Adipose tissue inflammation and metabolic syndrome. The proactive role of probiotics. Eur. J. Nutr. 2019, 58, 27–43. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metab. Clin. Exp. 2019, 92, 121–135. [Google Scholar] [CrossRef]
- Gacci, M.; Russo, G.I.; De Nunzio, C.; Sebastianelli, A.; Salvi, M.; Vignozzi, L.; Tubaro, A.; Morgia, G.; Serni, S. Meta-analysis of metabolic syndrome and prostate cancer. Prostate Cancer Prostatic Dis. 2017, 20, 146–155. [Google Scholar] [CrossRef]
- Duarte, M.F.; Luis, C.; Baylina, P.; Faria, M.I.; Fernandes, R.; La Fuente, J.M. Clinical and metabolic implications of obesity in prostate cancer: Is testosterone a missing link? Aging Male Off. J. Int. Soc. Study Aging Male 2019, 22, 228–240. [Google Scholar] [CrossRef]
- McGrowder, D.A.; Jackson, L.A.; Crawford, T.V. Prostate cancer and metabolic syndrome: Is there a link? Asian Pac. J. Cancer Prev. APJCP 2012, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Beebe-Dimmer, J.L.; Dunn, R.L.; Sarma, A.V.; Montie, J.E.; Cooney, K.A. Features of the metabolic syndrome and prostate cancer in African-American men. Cancer 2007, 109, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Beebe-Dimmer, J.L.; Nock, N.L.; Neslund-Dudas, C.; Rundle, A.; Bock, C.H.; Tang, D.; Jankowski, M.; Rybicki, B.A. Racial differences in risk of prostate cancer associated with metabolic syndrome. Urology 2009, 74, 185–190. [Google Scholar] [CrossRef]
- Choi, J.B.; Myong, J.P.; Lee, Y.; Koh, J.S.; Hong, S.H.; Yoon, B.I.; Ha, U.S. Impact of age and metabolic syndrome-like components on prostate cancer development: A nationwide population-based cohort study. Transl. Androl. Urol. 2021, 10, 2990–2997. [Google Scholar] [CrossRef]
- Hammarsten, J.; Damber, J.E.; Haghsheno, M.A.; Mellström, D.; Peeker, R. A stage-dependent link between metabolic syndrome components and incident prostate cancer. Nat. Rev. Urol. 2018, 15, 321–333. [Google Scholar] [CrossRef]
- Gao, X.; Li, R.; Jin, T.; Tang, H. The Association Between Metabolic Syndrome and Prostate Cancer Risk: A Large-Scale Investigation and Study of Chinese. Front. Endocrinol. 2022, 13, 787268. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Chiodini, P.; Capuano, A.; Bellastella, G.; Maiorino, M.I.; Rafaniello, C.; Giugliano, D. Metabolic syndrome and postmenopausal breast cancer: Systematic review and meta-analysis. Menopause 2013, 20, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CAA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Simon, R.J.; Bott, K.L.N. Prostate Cancer; Exon Publications: Brisbane, Australia, 2021. [Google Scholar] [CrossRef]
- Rawla, P. Epidemiology of prostate cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CAA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.P.; Costa, R.; Alves, M.G.; Soares, R.; Baylina, P.; Fernandes, R. The Impact of Metabolic Syndrome and Type 2 Diabetes Mellitus on Prostate Cancer. Front. Cell Dev. Biol. 2022, 10, 843458. [Google Scholar] [CrossRef] [PubMed]
- Schrecengost, R.; Knudsen, K.E. Molecular pathogenesis and progression of prostate cancer. Semin. Oncol. 2013, 40, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Ettinger, S.L.; Qu, S.; Xue, H.; Nabavi, N.; Choi, S.Y.C.; Bell, R.H.; Mo, F.; Haegert, A.M.; Gout, P.W.; et al. Metabolic heterogeneity signature of primary treatment-naïve prostate cancer. Oncotarget 2017, 8, 25928–25941. [Google Scholar] [CrossRef] [PubMed]
- Chetta, P.; Zadra, G. Metabolic reprogramming as an emerging mechanism of resistance to endocrine therapies in prostate cancer. Cancer Drug Resist. 2021, 4, 143–162. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CAA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef]
- Center, M.M.; Jemal, A.; Lortet-Tieulent, J.; Ward, E.; Ferlay, J.; Brawley, O.; Bray, F. International variation in prostate cancer incidence and mortality rates. Eur. Urol. 2012, 61, 1079–1092. [Google Scholar] [CrossRef]
- Esposito, K.; Chiodini, P.; Capuano, A.; Bellastella, G.; Maiorino, M.I.; Parretta, E.; Lenzi, A.; Giugliano, D. Effect of metabolic syndrome and its components on prostate cancer risk: Meta-analysis. J. Endocrinol. Investig. 2013, 36, 132–139. [Google Scholar] [CrossRef]
- Wolk, A.; Mantzoros, C.S.; Andersson, S.O.; Bergström, R.; Signorello, L.B.; Lagiou, P.; Adami, H.O.; Trichopoulos, D. Insulin-like growth factor 1 and prostate cancer risk: A population-based, case-control study. J. Natl. Cancer Inst. 1998, 90, 911–915. [Google Scholar] [CrossRef]
- Bhindi, B.; Locke, J.; Alibhai, S.M.H.; Kulkarni, G.S.; Margel, D.S.; Hamilton, R.J.; Finelli, A.; Trachtenberg, J.; Zlotta, A.R.; Toi, A.; et al. Dissecting the association between metabolic syndrome and prostate cancer risk: Analysis of a large clinical cohort. Eur. Urol. 2015, 67, 64–70. [Google Scholar] [CrossRef]
- Karzai, F.H.; Madan, R.A.; Dahut, W.L. Metabolic syndrome in prostate cancer: Impact on risk and outcomes. Future Oncol. 2016, 12, 1947–1955. [Google Scholar] [CrossRef]
- Conteduca, V.; Di Lorenzo, G.; Bozza, G.; Ardito, R.; Aieta, M. Metabolic syndrome as a peculiar target for management of prostate cancer patients. Clin. Genitourin. Cancer 2013, 11, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Laukkanen, J.A.; Laaksonen, D.E.; Niskanen, L.; Pukkala, E.; Hakkarainen, A.; Salonen, J.T. Metabolic Syndrome and the Risk of Prostate Cancer in Finnish Men: A Population-Based Study. Cancer Epidemiol. Biomark. Prev. 2004, 13, 1646–1650. [Google Scholar] [CrossRef]
- Blanc-Lapierre, A.; Spence, A.; Karakiewicz, P.I.; Aprikian, A.; Saad, F.; Parent, M. Metabolic syndrome and prostate cancer risk in a population-based case-control study in Montreal, Canada. BMC Public Health 2015, 15, 913. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, Q.; Chang, C.; Wang, X.; Xie, L. Metabolic Syndrome Is Not Associated With Prostate Cancer Recurrence: A Retrospective Analysis of a Chinese Cohort. Front. Oncol. 2020, 10, 63. [Google Scholar] [CrossRef]
- Tande, A.J.; Platz, E.A.; Folsom, A.R. The Metabolic Syndrome Is Associated with Reduced Risk of Prostate Cancer. Am. J. Epidemiol. 2006, 164, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Monroy-Iglesias, M.J.; Russell, B.; Crawley, D.; Allen, N.E.; Travis, R.C.; Perez-Cornago, A.; Van Hemelrijck, M.; Beckmann, K. Metabolic syndrome biomarkers and prostate cancer risk in the UK Biobank. Wiley Online Libr. 2021, 148, 825–834. [Google Scholar] [CrossRef]
- Grundmark, B.; Garmo, H.; Loda, M.; Busch, C.; Holmberg, L.; Zethelius, B. The Metabolic Syndrome and the Risk of Prostate Cancer under Competing Risks of Death from Other Causes. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2088–2096. [Google Scholar] [CrossRef]
- Martin, R.M.; Vatten, L.; Gunnell, D.; Romundstad, P.; Nilsen, T.I.L. Components of the metabolic syndrome and risk of prostate cancer: The HUNT 2 cohort, Norway. Cancer Causes Control. 2009, 20, 1181–1192. [Google Scholar] [CrossRef]
- Ford, E.S.; Giles, W.H.; Dietz, W.H. Prevalence of the metabolic syndrome among US adults: Findings from the third National Health and Nutrition Examination Survey. JAMA 2002, 287, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Sourbeer, K.N.; Howard, L.E.; Andriole, G.L.; Moreira, D.M.; Castro-Santamaria, R.; Freedland, S.J.; Vidal, A.C. Metabolic syndrome-like components and prostate cancer risk: Results from the Reduction by Dutasteride of Prostate Cancer Events (REDUCE) study. BJU Int. 2015, 115, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Lund Håheim, L.; Wisløff, T.F.; Holme, I.; Nafstad, P. Metabolic syndrome predicts prostate cancer in a cohort of middle-aged Norwegian men followed for 27 years. Am. J. Epidemiol. 2006, 164, 769–774. [Google Scholar] [CrossRef]
- Platz, E.A.; Till, C.; Goodman, P.J.; Parnes, H.L.; Figg, W.D.; Albanes, D.; Neuhouser, M.L.; Klein, E.A.; Thompson, I.M., Jr.; Kristal, A.R. Men with low serum cholesterol have a lower risk of high-grade prostate cancer in the placebo arm of the prostate cancer prevention trial. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2807–2813. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.M.; Stampfer, M.J.; Ma, J.; Gann, P.; Gaziano, J.M.; Pollak, M.; Giovannucci, E. Insulin-like growth factor-I (IGF-I) and IGF binding protein-3 as predictors of advanced-stage prostate cancer. J. Natl. Cancer Inst. 2002, 94, 1099–1106. [Google Scholar] [CrossRef]
- Lai, G.Y.; Giovannucci, E.L.; Pollak, M.N.; Peskoe, S.B.; Stampfer, M.J.; Willett, W.C.; Platz, E.A. Association of C-peptide and leptin with prostate cancer incidence in the Health Professionals Follow-up Study. Cancer Causes Control CCC 2014, 25, 625–632. [Google Scholar] [CrossRef]
- Li, H.; Stampfer, M.J.; Mucci, L.; Rifai, N.; Qiu, W.; Kurth, T.; Ma, J. A 25-year prospective study of plasma adiponectin and leptin concentrations and prostate cancer risk and survival. Clin. Chem. 2010, 56, 34–43. [Google Scholar] [CrossRef]
- Powell, I.J.; Bollig-Fischer, A. Minireview: The molecular and genomic basis for prostate cancer health disparities. Mol. Endocrinol. 2013, 27, 879–891. [Google Scholar] [CrossRef]
- Sfanos, K.S.; De Marzo, A.M. Prostate cancer and inflammation: The evidence. Histopathology 2012, 60, 199–215. [Google Scholar] [CrossRef]
- Tsilidis, K.K.; Helzlsouer, K.J.; Smith, M.W.; Grinberg, V.; Hoffman-Bolton, J.; Clipp, S.L.; Visvanathan, K.; Platz, E.A. Association of common polymorphisms in IL10, and in other genes related to inflammatory response and obesity with colorectal cancer. Cancer Causes Control CCC 2009, 20, 1739–1751. [Google Scholar] [CrossRef]
- Wallner, L.P.; Morgenstern, H.; McGree, M.E.; Jacobson, D.J.; St Sauver, J.L.; Jacobsen, S.J.; Sarma, A.V. The effects of metabolic conditions on prostate cancer incidence over 15 years of follow-up: Results from the Olmsted County Study. BJU Int. 2011, 107, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, Y.R.; Morag, O.; Benderly, M.; Boyko, V.; Novikov, I.; Dicker, A.P.; Goldbourt, U.; Behar, S.; Barchana, M.; Wolf, I. Association between metabolic syndrome, diabetes mellitus and prostate cancer risk. Prostate Cancer Prostatic Dis. 2013, 16, 181–186. [Google Scholar] [CrossRef]
- Ma, J.; Li, H.; Giovannucci, E.; Mucci, L.; Qiu, W.; Nguyen, P.L.; Gaziano, J.M.; Pollak, M.; Stampfer, M.J. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: A long-term survival analysis. Lancet. Oncol. 2008, 9, 1039–1047. [Google Scholar] [CrossRef]
- Zadra, G.; Loda, M. Metabolic Vulnerabilities of Prostate Cancer: Diagnostic and Therapeutic Opportunities. Cold Spring Harb. Perspect. Med. 2018, 8, a030569. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Dong, B.; Ning, J.; Shao, X.; Zhao, L.; Jiang, Q.; Ji, H.; Cai, A.; Xue, W.; Gao, H. NMR-based metabolomics analysis identifies discriminatory metabolic disturbances in tissue and biofluid samples for progressive prostate cancer. Clin. Chim. Acta 2020, 501, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Giskeødegård, G.F.; Bertilsson, H.; Selnæs, K.M.; Wright, A.J.; Bathen, T.F.; Viset, T.; Halgunset, J.; Angelsen, A.; Gribbestad, I.S.; Tessem, M.B. Spermine and citrate as metabolic biomarkers for assessing prostate cancer aggressiveness. PLoS ONE 2013, 8, e62375. [Google Scholar] [CrossRef]
- Gómez-Cebrián, N.; Poveda, J.L.; Pineda-Lucena, A.; Puchades-Carrasco, L. Metabolic Phenotyping in Prostate Cancer Using Multi-Omics Approaches. Cancers 2022, 14, 596. [Google Scholar] [CrossRef]
- Sanità, P.; Capulli, M.; Teti, A.; Galatioto, G.P.; Vicentini, C.; Chiarugi, P.; Bologna, M.; Angelucci, A. Tumor-stroma metabolic relationship based on lactate shuttle can sustain prostate cancer progression. BMC Cancer 2014, 14, 154. [Google Scholar] [CrossRef]
- Pertega-Gomes, N.; Felisbino, S.; Massie, C.E.; Vizcaino, J.R.; Coelho, R.; Sandi, C.; Simoes-Sousa, S.; Jurmeister, S.; Ramos-Montoya, A.; Asim, M.; et al. A glycolytic phenotype is associated with prostate cancer progression and aggressiveness: A role for monocarboxylate transporters as metabolic targets for therapy. J. Pathol. 2015, 236, 517–530. [Google Scholar] [CrossRef]
- Pan, T.; Gao, L.; Wu, G.; Shen, G.; Xie, S.; Wen, H.; Yang, J.; Zhou, Y.; Tu, Z.; Qian, W. Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochem. Biophys. Res. Commun. 2015, 456, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hardie, R.A.; Hoy, A.J.; van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Feun, L.; You, M.; Wu, C.J.; Kuo, M.T.; Wangpaichitr, M.; Spector, S.; Savaraj, N. Arginine deprivation as a targeted therapy for cancer. Curr. Pharm. Des. 2008, 14, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.E. Androgen resistance--the clinical and molecular spectrum. N. Engl. J. Med. 1992, 326, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.R.; Pinto, J.; Amaro, F.; Bastos, M.L.; Carvalho, M.; Guedes de Pinho, P. Advances and Perspectives in Prostate Cancer Biomarker Discovery in the Last 5 Years through Tissue and Urine Metabolomics. Metabolites 2021, 11, 181. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Oh, S.; Jin, X.; An, Y.J.; Park, S. Cancer metabolomics in basic science perspective. Arch. Pharmacal Res. 2015, 38, 372–380. [Google Scholar] [CrossRef]
- Cutruzzolà, F.; Giardina, G.; Marani, M.; Macone, A.; Paiardini, A.; Rinaldo, S.; Paone, A. Glucose Metabolism in the Progression of Prostate Cancer. Front. Physiol. 2017, 8, 97. [Google Scholar] [CrossRef]
- Cardoso, H.J.; Carvalho, T.M.A.; Fonseca, L.R.S.; Figueira, M.I.; Vaz, C.V.; Socorro, S. Revisiting prostate cancer metabolism: From metabolites to disease and therapy. Med. Res. Rev. 2021, 41, 1499–1538. [Google Scholar] [CrossRef]
- Green, S.M.; Mostaghel, E.A.; Nelson, P.S. Androgen action and metabolism in prostate cancer. Mol. Cell. Endocrinol. 2012, 360, 3–13. [Google Scholar] [CrossRef]
- Ross, R.K.; Pike, M.C.; Coetzee, G.A.; Reichardt, J.K.; Yu, M.C.; Feigelson, H.; Stanczyk, F.Z.; Kolonel, L.N.; Henderson, B.E. Androgen metabolism and prostate cancer: Establishing a model of genetic susceptibility. Cancer Res. 1998, 58, 4497–4504. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Detterbeck, F.C.; Falen, S.; Rivera, M.P.; Halle, J.S.; Socinski, M.A. Seeking a home for a PET, part 2: Defining the appropriate place for positron emission tomography imaging in the staging of patients with suspected lung cancer. Chest 2004, 125, 2300–2308. [Google Scholar] [CrossRef]
- Peyruchaud, O.; Serre, C.M.; NicAmhlaoibh, R.; Fournier, P.; Clezardin, P. Angiostatin inhibits bone metastasis formation in nude mice through a direct anti-osteoclastic activity. J. Biol. Chem. 2003, 278, 45826–45832. [Google Scholar] [CrossRef] [PubMed]
- Peyruchaud, O.; Serre, C.M.; NicAmhlaoibh, R.; Sveigaard, C.; Clézardin, P. Does tumor angiogenesis play a role in bone metastatic process? Rev. Med. Suisse Rom. 2004, 124, 83–84. [Google Scholar] [PubMed]
- Staller, P.; Sulitkova, J.; Lisztwan, J.; Moch, H.; Oakeley, E.J.; Krek, W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature 2003, 425, 307–311. [Google Scholar] [CrossRef] [PubMed]
- LaTulippe, E.; Satagopan, J.; Smith, A.; Scher, H.; Scardino, P.; Reuter, V.; Gerald, W.L. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002, 62, 4499–4506. [Google Scholar]
- Riley, D.E.; Krieger, J.N. Short tandem repeat polymorphism linkage to the androgen receptor gene in prostate carcinoma. Cancer 2001, 92, 2603–2608. [Google Scholar] [CrossRef]
- Chen, G.; Gharib, T.G.; Wang, H.; Huang, C.C.; Kuick, R.; Thomas, D.G.; Shedden, K.A.; Misek, D.E.; Taylor, J.M.; Giordano, T.J.; et al. Protein profiles associated with survival in lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2003, 100, 13537–13542. [Google Scholar] [CrossRef] [PubMed]
- Migita, T.; Oda, Y.; Naito, S.; Morikawa, W.; Kuwano, M.; Tsuneyoshi, M. The accumulation of angiostatin-like fragments in human prostate carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2001, 7, 2750–2756. [Google Scholar]
- Daly, E.B.; Wind, T.; Jiang, X.M.; Sun, L.; Hogg, P.J. Secretion of phosphoglycerate kinase from tumour cells is controlled by oxygen-sensing hydroxylases. Biochim. Biophys. Acta 2004, 1691, 17–22. [Google Scholar] [CrossRef]
- Lay, A.J.; Jiang, X.M.; Kisker, O.; Flynn, E.; Underwood, A.; Condron, R.; Hogg, P.J. Phosphoglycerate kinase acts in tumour angiogenesis as a disulphide reductase. Nature 2000, 408, 869–873. [Google Scholar] [CrossRef]
- Zhang, D.; Tai, L.K.; Wong, L.L.; Chiu, L.L.; Sethi, S.K.; Koay, E.S. Proteomic study reveals that proteins involved in metabolic and detoxification pathways are highly expressed in HER-2/neu-positive breast cancer. Mol. Cell. Proteom. MCP 2005, 4, 1686–1696. [Google Scholar] [CrossRef]
- Hwang, T.L.; Liang, Y.; Chien, K.Y.; Yu, J.S. Overexpression and elevated serum levels of phosphoglycerate kinase 1 in pancreatic ductal adenocarcinoma. Proteomics 2006, 6, 2259–2272. [Google Scholar] [CrossRef]
- Diedrich, J.D.; Rajagurubandara, E.; Herroon, M.K.; Mahapatra, G.; Hüttemann, M.; Podgorski, I. Bone marrow adipocytes promote the Warburg phenotype in metastatic prostate tumors via HIF-1α activation. Oncotarget 2016, 7, 64854–64877. [Google Scholar] [CrossRef]
- Vaughan, M. The production and release of glycerol by adipose tissue incubated in vitro. J. Biol. Chem. 1962, 237, 3354–3358. [Google Scholar] [CrossRef] [PubMed]
- Langin, D. Control of fatty acid and glycerol release in adipose tissue lipolysis. Comptes Rendus Biol. 2006, 329, 598–607. [Google Scholar] [CrossRef]
- Maeda, N.; Funahashi, T.; Shimomura, I. Metabolic impact of adipose and hepatic glycerol channels aquaporin 7 and aquaporin 9. Nat. Clin. Pract. Endocrinol. Metab. 2008, 4, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Bezwada, D.; Zandbergen, M.; Greco, F.F.; Chiang, C.-Y.; Tasdemir, M.; Fahrmann, J.; Grapov, D.; La Frano, M.R.; Vu, H.S. Adipocytes reprogram glucose metabolism in cancer cells promoting metastasis. bioRxiv 2022, 10. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. A comprehensive review of the role of zinc in normal prostate function and metabolism; and its implications in prostate cancer. Arch. Biochem. Biophys. 2016, 611, 100–112. [Google Scholar] [CrossRef]
- Schöpf, B.; Weissensteiner, H.; Schäfer, G.; Fazzini, F.; Charoentong, P.; Naschberger, A.; Rupp, B.; Fendt, L.; Bukur, V.; Giese, I.; et al. OXPHOS remodeling in high-grade prostate cancer involves mtDNA mutations and increased succinate oxidation. Nat. Commun. 2020, 11, 1487. [Google Scholar] [CrossRef]
- Weber, A.; Klocker, H.; Oberacher, H.; Gnaiger, E.; Neuwirt, H.; Sampson, N.; Eder, I.E. Succinate Accumulation Is Associated with a Shift of Mitochondrial Respiratory Control and HIF-1α Upregulation in PTEN Negative Prostate Cancer Cells. Int. J. Mol. Sci. 2018, 19, 2129. [Google Scholar] [CrossRef] [PubMed]
- Bader, D.A.; Hartig, S.M.; Putluri, V.; Foley, C.; Hamilton, M.P.; Smith, E.A.; Saha, P.K.; Panigrahi, A.; Walker, C.; Zong, L.; et al. Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nat. Metab. 2019, 1, 70–85. [Google Scholar] [CrossRef]
- Zadra, G.; Photopoulos, C.; Tyekucheva, S.; Heidari, P.; Weng, Q.P.; Fedele, G.; Liu, H.; Scaglia, N.; Priolo, C.; Sicinska, E.; et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 2014, 6, 519–538. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Fraenkel, D.G.; Horecker, B.L. Fructose-1, 6-diphosphatase and acid hexose phosphatase of Escherichia coli. J. Bacteriol. 1965, 90, 837–842. [Google Scholar] [CrossRef]
- Valdés-Hevia, M.D.; de la Guerra, R.; Gancedo, C. Isolation and characterization of the gene encoding phosphoenolpyruvate carboxykinase from Saccharomyces cerevisiae. FEBS Lett. 1989, 258, 313–316. [Google Scholar] [CrossRef]
- Zhao, J.; Li, J.; Fan, T.W.; Hou, S.X. Glycolytic reprogramming through PCK2 regulates tumor initiation of prostate cancer cells. Oncotarget 2017, 8, 83602. [Google Scholar] [CrossRef]
- Grasmann, G.; Smolle, E.; Olschewski, H.; Leithner, K. Gluconeogenesis in cancer cells—Repurposing of a starvation-induced metabolic pathway? Biochim. Biophys. Acta. Rev. Cancer 2019, 1872, 24–36. [Google Scholar] [CrossRef]
- Freedland, S.J.; Aronson, W.J. Obesity and prostate cancer. Urology 2005, 65, 433–439. [Google Scholar] [CrossRef]
- Clarke, N.W.; Brown, M.D. The influence of lipid metabolism on prostate cancer development and progression: Is it time for a closer look? Eur. Urol. 2007, 52, 3–4. [Google Scholar] [CrossRef]
- Suburu, J.; Chen, Y.Q. Lipids and prostate cancer. Prostaglandins Other Lipid Mediat. 2012, 98, 1–10. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Galleggiante, V.; Giglio, A.; Palazzo, S.; Ferro, M.; Simone, C.; Bettocchi, C.; Battaglia, M.; Ditonno, P. Metabolomic profiling for the identification of novel diagnostic markers in prostate cancer. Expert Rev. Mol. Diagn. 2015, 15, 1211–1224. [Google Scholar] [CrossRef] [PubMed]
- Ferro, M.; Terracciano, D.; Buonerba, C.; Lucarelli, G.; Bottero, D.; Perdonà, S.; Autorino, R.; Serino, A.; Cantiello, F.; Damiano, R.; et al. The emerging role of obesity, diet and lipid metabolism in prostate cancer. Future Oncol. 2017, 13, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Daniels, G.; Lee, P.; Monaco, M.E. Lipid metabolism in prostate cancer. Am. J. Clin. Exp. Urol. 2014, 2, 111. [Google Scholar]
- Liu, Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006, 9, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, M.D.; Yevglevskis, M.; Lee, G.L.; Wood, P.J.; Threadgill, M.D.; Woodman, T.J. α-Methylacyl-CoA racemase (AMACR): Metabolic enzyme, drug metabolizer and cancer marker P504S. Prog. Lipid Res. 2013, 52, 220–230. [Google Scholar] [CrossRef]
- Huang, W.C.; Li, X.; Liu, J.; Lin, J.; Chung, L.W. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol. Cancer Res. MCR 2012, 10, 133–142. [Google Scholar] [CrossRef]
- Swinnen, J.V.; Heemers, H.; van de Sande, T.; de Schrijver, E.; Brusselmans, K.; Heyns, W.; Verhoeven, G. Androgens, lipogenesis and prostate cancer. J. Steroid Biochem. Mol. Biol. 2004, 92, 273–279. [Google Scholar] [CrossRef]
- Ettinger, S.L.; Sobel, R.; Whitmore, T.G.; Akbari, M.; Bradley, D.R.; Gleave, M.E.; Nelson, C.C. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res. 2004, 64, 2212–2221. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Li, P.; Cheng, C.; Zhao, Y.; Li, D.; Du, C. Cholesterol Levels in Blood and the Risk of Prostate Cancer: A Meta-analysis of 14 Prospective Studies. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1086–1093. [Google Scholar] [CrossRef]
- Likus, W.; Siemianowicz, K.; Bieńk, K.; Pakuła, M.; Pathak, H.; Dutta, C.; Wang, Q.; Shojaei, S.; Assaraf, Y.G.; Ghavami, S.; et al. Could drugs inhibiting the mevalonate pathway also target cancer stem cells? Drug Resist. Updat. 2016, 25, 13–25. [Google Scholar] [CrossRef]
- Matsushita, Y.; Nakagawa, H.; Koike, K. Lipid Metabolism in Oncology: Why It Matters, How to Research, and How to Treat. Cancers 2021, 13, 474. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J. Cholesterol, statins and cancer. Clin. Exp. Pharmacol. Physiol. 2007, 34, 135–141. [Google Scholar] [CrossRef]
- Lewis, C.A.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D.; et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140. [Google Scholar] [CrossRef]
- Pelton, K.; Freeman, M.R.; Solomon, K.R. Cholesterol and prostate cancer. Curr. Opin. Pharmacol. 2012, 12, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Howe, V.; Sharpe, L.J.; Alexopoulos, S.J.; Kunze, S.V.; Chua, N.K.; Li, D.; Brown, A.J. Cholesterol homeostasis: How do cells sense sterol excess? Chem. Phys. Lipids 2016, 199, 170–178. [Google Scholar] [CrossRef]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Schörghofer, D.; Kinslechner, K.; Preitschopf, A.; Schütz, B.; Röhrl, C.; Hengstschläger, M.; Stangl, H.; Mikula, M. The HDL receptor SR-BI is associated with human prostate cancer progression and plays a possible role in establishing androgen independence. Reprod. Biol. Endocrinol. RBE 2015, 13, 88. [Google Scholar] [CrossRef]
- Alioui, A.; Celhay, O.; Baron, S.; Lobaccaro, J.-M.A. Lipids and prostate cancer adenocarcinoma. Clin. Lipidol. 2014, 9, 643–655. [Google Scholar] [CrossRef]
- Jamnagerwalla, J.; Howard, L.E.; Allott, E.H.; Vidal, A.C.; Moreira, D.M.; Castro-Santamaria, R.; Andriole, G.L.; Freeman, M.R.; Freedland, S.J. Serum cholesterol and risk of high-grade prostate cancer: Results from the reduce study. Prostate Cancer Prostatic Dis. 2018, 21, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Sankanagoudar, S.; Dogra, P.N.; Mathur, S.R.; Chandra, N.C. A study on lipid profile in prostate carcinoma patients admitted in AIIMS, New Delhi. J Biomed Pharm Res 2014, 3, 49–51. [Google Scholar]
- Stopsack, K.H.; Gerke, T.A.; Andrén, O.; Andersson, S.O.; Giovannucci, E.L.; Mucci, L.A.; Rider, J.R. Cholesterol uptake and regulation in high-grade and lethal prostate cancers. Carcinogenesis 2017, 38, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Lethongsavarn, V.; Pinault, M.; Diedhiou, A.; Guimaraes, C.; Guibon, R.; Bruyère, F.; Mathieu, R.; Rioux-Leclercq, N.; Multigner, L.; Brureau, L.; et al. Tissue cholesterol metabolism and prostate cancer aggressiveness: Ethno-geographic variations. Prostate 2021, 81, 1365–1373. [Google Scholar] [CrossRef]
- Mostaghel, E.A.; Solomon, K.R.; Pelton, K.; Freeman, M.R.; Montgomery, R.B. Impact of circulating cholesterol levels on growth and intratumoral androgen concentration of prostate tumors. PLoS ONE 2012, 7, e30062. [Google Scholar] [CrossRef]
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resistant tumor growth. Cancer Res. 2008, 68, 4447–4454. [Google Scholar] [CrossRef]
- Hager, M.H.; Solomon, K.R.; Freeman, M.R. The role of cholesterol in prostate cancer. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 379–385. [Google Scholar] [CrossRef]
- Platz, E.A.; Clinton, S.K.; Giovannucci, E. Association between plasma cholesterol and prostate cancer in the PSA era. Int. J. Cancer 2008, 123, 1693–1698. [Google Scholar] [CrossRef]
- Leon, C.G.; Locke, J.A.; Adomat, H.H.; Etinger, S.L.; Twiddy, A.L.; Neumann, R.D.; Nelson, C.C.; Guns, E.S.; Wasan, K.M. Alterations in cholesterol regulation contribute to the production of intratumoral androgens during progression to castration-resistant prostate cancer in a mouse xenograft model. Prostate 2010, 70, 390–400. [Google Scholar] [CrossRef]
- Zhuang, L.; Lin, J.; Lu, M.L.; Solomon, K.R.; Freeman, M.R. Cholesterol-rich lipid rafts mediate akt-regulated survival in prostate cancer cells. Cancer Res. 2002, 62, 2227–2231. [Google Scholar]
- Twiddy, A.L.; Cox, M.E.; Wasan, K.M. Knockdown of scavenger receptor class B type I reduces prostate specific antigen secretion and viability of prostate cancer cells. Prostate 2012, 72, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Stopsack, K.H.; Gerke, T.A.; Sinnott, J.A.; Penney, K.L.; Tyekucheva, S.; Sesso, H.D.; Andersson, S.O.; Andrén, O.; Cerhan, J.R.; Giovannucci, E.L.; et al. Cholesterol Metabolism and Prostate Cancer Lethality. Cancer Res. 2016, 76, 4785–4790. [Google Scholar] [CrossRef] [PubMed]
- Farwell, W.R.; D’Avolio, L.W.; Scranton, R.E.; Lawler, E.V.; Gaziano, J.M. Statins and prostate cancer diagnosis and grade in a veterans population. J. Natl. Cancer Inst. 2011, 103, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Yu, O.; Eberg, M.; Benayoun, S.; Aprikian, A.; Batist, G.; Suissa, S.; Azoulay, L. Use of statins and the risk of death in patients with prostate cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 5–11. [Google Scholar] [CrossRef]
- Sanguinetti, A.R.; Mastick, C.C. c-Abl is required for oxidative stress-induced phosphorylation of caveolin-1 on tyrosine 14. Cell. Signal. 2003, 15, 289–298. [Google Scholar] [CrossRef]
- Sanguinetti, A.R.; Cao, H.; Corley Mastick, C. Fyn is required for oxidative- and hyperosmotic-stress-induced tyrosine phosphorylation of caveolin-1. Biochem. J. 2003, 376, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Nomura, R.; Fujimoto, T. Tyrosine-phosphorylated caveolin-1: Immunolocalization and molecular characterization. Mol. Biol. Cell 1999, 10, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Hau, A.M.; Gupta, S.; Leivo, M.Z.; Nakashima, K.; Macias, J.; Zhou, W.; Hodge, A.; Wulfkuhle, J.; Conkright, B.; Bhuvaneshwar, K.; et al. Dynamic Regulation of Caveolin-1 Phosphorylation and Caveolae Formation by Mammalian Target of Rapamycin Complex 2 in Bladder Cancer Cells. Am. J. Pathol. 2019, 189, 1846–1862. [Google Scholar] [CrossRef]
- Orlichenko, L.; Huang, B.; Krueger, E.; McNiven, M.A. Epithelial growth factor-induced phosphorylation of caveolin 1 at tyrosine 14 stimulates caveolae formation in epithelial cells. J. Biol. Chem. 2006, 281, 4570–4579. [Google Scholar] [CrossRef]
- Cheng, J.P.X.; Nichols, B.J. Caveolae: One Function or Many? Trends Cell Biol. 2016, 26, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Caveolae and signalling in cancer. Nat. Rev. Cancer 2015, 15, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Frank, P.G.; Woodman, S.E.; Park, D.S.; Lisanti, M.P. Caveolin, caveolae, and endothelial cell function. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Tahir, S.A.; Yang, G.; Goltsov, A.; Song, K.D.; Ren, C.; Wang, J.; Chang, W.; Thompson, T.C. Caveolin-1-LRP6 signaling module stimulates aerobic glycolysis in prostate cancer. Cancer Res. 2013, 73, 1900–1911. [Google Scholar] [CrossRef] [PubMed]
- Shatz, M.; Liscovitch, M. Caveolin-1: A tumor-promoting role in human cancer. Int. J. Radiat. Biol. 2008, 84, 177–189. [Google Scholar] [CrossRef]
- Shankar, J.; Boscher, C.; Nabi, I.R. Caveolin-1, galectin-3 and lipid raft domains in cancer cell signalling. Essays Biochem. 2015, 57, 189–201. [Google Scholar] [CrossRef]
- Burgermeister, E.; Liscovitch, M.; Röcken, C.; Schmid, R.M.; Ebert, M.P. Caveats of caveolin-1 in cancer progression. Cancer Lett. 2008, 268, 187–201. [Google Scholar] [CrossRef]
- Mi, L.; Zhu, F.; Yang, X.; Lu, J.; Zheng, Y.; Zhao, Q.; Wen, X.; Lu, A.; Wang, M.; Zheng, M.; et al. The metastatic suppressor NDRG1 inhibits EMT, migration and invasion through interaction and promotion of caveolin-1 ubiquitylation in human colorectal cancer cells. Oncogene 2017, 36, 4323–4335. [Google Scholar] [CrossRef] [PubMed]
- Hehlgans, S.; Cordes, N. Caveolin-1: An essential modulator of cancer cell radio-and chemoresistance. Am. J. Cancer Res. 2011, 1, 521–530. [Google Scholar]
- Raudenska, M.; Gumulec, J.; Balvan, J.; Masarik, M. Caveolin-1 in oncogenic metabolic symbiosis. Int. J. Cancer 2020, 147, 1793–1807. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Newmeyer, D.D.; Ferguson-Miller, S. Mitochondria: Releasing power for life and unleashing the machineries of death. Cell 2003, 112, 481–490. [Google Scholar] [CrossRef]
- Moindjie, H.; Rodrigues-Ferreira, S.; Nahmias, C. Mitochondrial Metabolism in Carcinogenesis and Cancer Therapy. Cancers 2021, 13, 3311. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.G.; Ghiraldeli, L.P.; Pardee, T.S. Mitochondria in cancer metabolism, an organelle whose time has come? Biochim. Biophys. Acta. Rev. Cancer 2018, 1870, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Sainero-Alcolado, L.; Liaño-Pons, J.; Ruiz-Pérez, M.V.; Arsenian-Henriksson, M. Targeting mitochondrial metabolism for precision medicine in cancer. Cell Death Differ. 2022, 29, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Blando, J.; Perez, C.J.; Wang, H.; Benavides, F.J.; Kazanietz, M.G. Activation of nuclear factor κB (NF-κB) in prostate cancer is mediated by protein kinase C epsilon (PKCepsilon). J. Biol. Chem. 2012, 287, 37570–37582. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Uzquiza, A.; Lopez-Haber, C.; Jernigan, D.L.; Fatatis, A.; Kazanietz, M.G. PKCε Is an Essential Mediator of Prostate Cancer Bone Metastasis. Mol. Cancer Res. MCR 2015, 13, 1336–1346. [Google Scholar] [CrossRef] [PubMed]
- Longoni, N.; Sarti, M.; Albino, D.; Civenni, G.; Malek, A.; Ortelli, E.; Pinton, S.; Mello-Grand, M.; Ostano, P.; D’Ambrosio, G.; et al. ETS transcription factor ESE1/ELF3 orchestrates a positive feedback loop that constitutively activates NF-κB and drives prostate cancer progression. Cancer Res. 2013, 73, 4533–4547. [Google Scholar] [CrossRef]
- Nadiminty, N.; Lou, W.; Sun, M.; Chen, J.; Yue, J.; Kung, H.J.; Evans, C.P.; Zhou, Q.; Gao, A.C. Aberrant activation of the androgen receptor by NF-kappaB2/p52 in prostate cancer cells. Cancer Res. 2010, 70, 3309–3319. [Google Scholar] [CrossRef]
- Jeong, J.H.; Park, S.J.; Dickinson, S.I.; Luo, J.L. A Constitutive Intrinsic Inflammatory Signaling Circuit Composed of miR-196b, Meis2, PPP3CC, and p65 Drives Prostate Cancer Castration Resistance. Mol. Cell 2017, 65, 154–167. [Google Scholar] [CrossRef]
- Wang, J.; Yi, S.; Zhou, J.; Zhang, Y.; Guo, F. The NF-κB subunit RelB regulates the migration and invasion abilities and the radio-sensitivity of prostate cancer cells. Int. J. Oncol. 2016, 49, 381–392. [Google Scholar] [CrossRef]
- Zhu, H.C.; Qiu, T.; Dan, C.; Liu, X.H.; Hu, C.H. Blockage of RelB expression by gene silencing enhances the radiosensitivity of androgen-independent prostate cancer cells. Mol. Med. Rep. 2015, 11, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Caino, M.C.; Kazanietz, M.G. Regulation of Transcriptional Networks by PKC Isozymes: Identification of c-Rel as a Key Transcription Factor for PKC-Regulated Genes. PLoS ONE 2013, 8, e67319. [Google Scholar] [CrossRef]
- Ahlqvist, K.; Saamarthy, K.; Syed Khaja, A.S.; Bjartell, A.; Massoumi, R. Expression of Id proteins is regulated by the Bcl-3 proto-oncogene in prostate cancer. Oncogene 2013, 32, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, V.K.; Studer, L.; Gerald, W.; Socci, N.D.; Scher, H.I. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-κB signalling. Nat. Commun. 2011, 2, 162. [Google Scholar] [CrossRef]
- Nunes, J.J.; Pandey, S.K.; Yadav, A.; Goel, S.; Ateeq, B. Targeting NF-kappa B Signaling by Artesunate Restores Sensitivity of Castrate-Resistant Prostate Cancer Cells to Antiandrogens. Neoplasia 2017, 19, 333–345. [Google Scholar] [CrossRef]
- Jin, R.; Yamashita, H.; Yu, X.; Wang, J.; Franco, O.E.; Wang, Y.; Hayward, S.W.; Matusik, R.J. Inhibition of NF-kappa B signaling restores responsiveness of castrate-resistant prostate cancer cells to anti-androgen treatment by decreasing androgen receptor-variant expression. Oncogene 2015, 34, 3700–3710. [Google Scholar] [CrossRef]
- Jin, R.J.; Lho, Y.; Connelly, L.; Wang, Y.; Yu, X.; Saint Jean, L.; Case, T.C.; Ellwood-Yen, K.; Sawyers, C.L.; Bhowmick, N.A.; et al. The nuclear factor-kappaB pathway controls the progression of prostate cancer to androgen-independent growth. Cancer Res. 2008, 68, 6762–6769. [Google Scholar] [CrossRef]
- Viljoen, T.C.; van Aswegen, C.H.; du Plessis, D.J. Influence of acetylsalicylic acid and metabolites on DU-145 prostatic cancer cell proliferation. Oncology 1995, 52, 465–469. [Google Scholar] [CrossRef]
- Xu, S.; Zhou, W.; Ge, J.; Zhang, Z. Prostaglandin E2 receptor EP4 is involved in the cell growth and invasion of prostate cancer via the cAMP-PKA/PI3K-Akt signaling pathway. Mol. Med. Rep. 2018, 17, 4702–4712. [Google Scholar] [CrossRef]
- Cai, Y.; Lee, Y.F.; Li, G.; Liu, S.; Bao, B.Y.; Huang, J.; Hsu, C.L.; Chang, C. A new prostate cancer therapeutic approach: Combination of androgen ablation with COX-2 inhibitor. Int. J. Cancer 2008, 123, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.J.; Lan, S.W.; Lu, Y.C.; Cheng, T.S.; Lai, P.F.; Tsai, C.H.; Hsu, T.W.; Lin, H.Y.; Shyu, H.Y.; Wu, S.R.; et al. Inhibition of cyclooxygenase-2-mediated matriptase activation contributes to the suppression of prostate cancer cell motility and metastasis. Oncogene 2017, 36, 4597–4609. [Google Scholar] [CrossRef] [PubMed]
- Klessig, D.F.; Tian, M.; Choi, H.W. Multiple Targets of Salicylic Acid and Its Derivatives in Plants and Animals. Front. Immunol. 2016, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shao, S.; Han, D.; Xu, Y.; Jiao, D.; Wu, J.; Yang, F.; Ge, Y.; Shi, S.; Li, Y.; et al. High mobility group box 1 promotes the epithelial-to-mesenchymal transition in prostate cancer PC3 cells via the RAGE/NF-κB signaling pathway. Int. J. Oncol. 2018, 53, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.W.; Tian, M.; Song, F.; Venereau, E.; Preti, A.; Park, S.W.; Hamilton, K.; Swapna, G.V.; Manohar, M.; Moreau, M.; et al. Aspirin’s Active Metabolite Salicylic Acid Targets High Mobility Group Box 1 to Modulate Inflammatory Responses. Mol. Med. 2015, 21, 526–535. [Google Scholar] [CrossRef]
- Ma, Y.; Brusselaers, N. Maintenance use of aspirin or other non-steroidal anti-inflammatory drugs (NSAIDs) and prostate cancer risk. Prostate Cancer Prostatic Dis. 2018, 21, 147–152. [Google Scholar] [CrossRef]
- Lessard, L.; Saad, F.; Le Page, C.; Diallo, J.S.; Péant, B.; Delvoye, N.; Mes-Masson, A.M. NF-kappaB2 processing and p52 nuclear accumulation after androgenic stimulation of LNCaP prostate cancer cells. Cell. Signal. 2007, 19, 1093–1100. [Google Scholar] [CrossRef]
- Campa, V.M.; Baltziskueta, E.; Bengoa-Vergniory, N.; Gorroño-Etxebarria, I.; Wesołowski, R.; Waxman, J.; Kypta, R.M. A screen for transcription factor targets of glycogen synthase kinase-3 highlights an inverse correlation of NFκB and androgen receptor signaling in prostate cancer. Oncotarget 2014, 5, 8173–8187. [Google Scholar] [CrossRef]
- Cui, Y.; Nadiminty, N.; Liu, C.; Lou, W.; Schwartz, C.T.; Gao, A.C. Upregulation of glucose metabolism by NF-κB2/p52 mediates enzalutamide resistance in castration-resistant prostate cancer cells. Endocr.-Relat. Cancer 2014, 21, 435–442. [Google Scholar] [CrossRef]
- Nadiminty, N.; Tummala, R.; Liu, C.; Lou, W.; Evans, C.P.; Gao, A.C. NF-κB2/p52:c-Myc:hnRNPA1 Pathway Regulates Expression of Androgen Receptor Splice Variants and Enzalutamide Sensitivity in Prostate Cancer. Mol. Cancer Ther. 2015, 14, 1884–1895. [Google Scholar] [CrossRef]
- Nadiminty, N.; Tummala, R.; Liu, C.; Yang, J.; Lou, W.; Evans, C.P.; Gao, A.C. NF-κB2/p52 induces resistance to enzalutamide in prostate cancer: Role of androgen receptor and its variants. Mol. Cancer Ther. 2013, 12, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Jain, G.; Voogdt, C.; Tobias, A.; Spindler, K.D.; Möller, P.; Cronauer, M.V.; Marienfeld, R.B. IκB kinases modulate the activity of the androgen receptor in prostate carcinoma cell lines. Neoplasia 2012, 14, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Staal, J.; Beyaert, R. Inflammation and NF-κB Signaling in Prostate Cancer: Mechanisms and Clinical Implications. Cells 2018, 7, 122. [Google Scholar] [CrossRef]
- Zhang, J.; Jung, Y.Y.; Mohan, C.D.; Deivasigamani, A.; Chinnathambi, A.; Alharbi, S.A.; Rangappa, K.S.; Hui, K.M.; Sethi, G.; Ahn, K.S. Nimbolide enhances the antitumor effect of docetaxel via abrogation of the NF-κB signaling pathway in prostate cancer preclinical models. Biochim. Biophys. Acta. Mol. Cell Res. 2022, 1869, 119344. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sun, S.; Zhou, C.; Sun, Z.; Wang, Q.; Sun, C. In vitro and in vivo anticancer activity of Lycorine in prostate cancer by inhibiting NF-κB signaling pathway. J. Cancer 2022, 13, 3151–3159. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, E.V.; Gavriliuk, L.A.; Pokrovsky, V.S. Oxidative Stress and Redox-Dependent Signaling in Prostate Cancer. Biochem. Biokhimiia 2022, 87, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Khurana, N.; Sikka, S.C. Targeting Crosstalk between Nrf-2, NF-κB and Androgen Receptor Signaling in Prostate Cancer. Cancers 2018, 10, 352. [Google Scholar] [CrossRef]
- Mukha, A.; Kahya, U.; Linge, A.; Chen, O.; Löck, S.; Lukiyanchuk, V.; Richter, S.; Alves, T.C.; Peitzsch, M.; Telychko, V.; et al. GLS-driven glutamine catabolism contributes to prostate cancer radiosensitivity by regulating the redox state, stemness and ATG5-mediated autophagy. Theranostics 2021, 11, 7844–7868. [Google Scholar] [CrossRef]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef]
- Kodama, M.; Oshikawa, K.; Shimizu, H.; Yoshioka, S.; Takahashi, M.; Izumi, Y.; Bamba, T.; Tateishi, C.; Tomonaga, T.; Matsumoto, M.; et al. A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer. Nat. Commun. 2020, 11, 1320. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 749. [Google Scholar] [CrossRef] [PubMed]
- Cruzat, V.; Macedo Rogero, M.; Noel Keane, K.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Shi, J.; Qin, X.; Zheng, Z.; Chen, M.; Lin, Z.; Ye, J.; Li, M. Hormone-Glutamine Metabolism: A Critical Regulatory Axis in Endocrine-Related Cancers. Int. J. Mol. Sci. 2022, 23, 10086. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef]
- Sun, J.; Bok, R.A.; DeLos Santos, J.; Upadhyay, D.; DeLos Santos, R.; Agarwal, S.; Van Criekinge, M.; Vigneron, D.B.; Aggarwal, R.; Peehl, D.M.; et al. Resistance to Androgen Deprivation Leads to Altered Metabolism in Human and Murine Prostate Cancer Cell and Tumor Models. Metabolites 2021, 11, 139. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Vayalil, P.K.; Landar, A. Mitochondrial oncobioenergetic index: A potential biomarker to predict progression from indolent to aggressive prostate cancer. Oncotarget 2015, 6, 43065–43080. [Google Scholar] [CrossRef]
- Li, T.; Copeland, C.; Le, A. Glutamine Metabolism in Cancer. Adv. Exp. Med. Biol. 2021, 1311, 17–38. [Google Scholar] [CrossRef]
- Xiao, D.; Zeng, L.; Yao, K.; Kong, X.; Wu, G.; Yin, Y. The glutamine-alpha-ketoglutarate (AKG) metabolism and its nutritional implications. Amino Acids 2016, 48, 2067–2080. [Google Scholar] [CrossRef]
- Ather, M.H.; Abbas, F.; Faruqui, N.; Israr, M.; Pervez, S. Correlation of three immunohistochemically detected markers of neuroendocrine differentiation with clinical predictors of disease progression in prostate cancer. BMC Urol. 2008, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Reina-Campos, M.; Linares, J.F.; Duran, A.; Cordes, T.; L’Hermitte, A.; Badur, M.G.; Bhangoo, M.S.; Thorson, P.K.; Richards, A.; Rooslid, T.; et al. Increased Serine and One-Carbon Pathway Metabolism by PKCλ/ι Deficiency Promotes Neuroendocrine Prostate Cancer. Cancer Cell 2019, 35, 385–400.e389. [Google Scholar] [CrossRef]
- Kareta, M.S.; Gorges, L.L.; Hafeez, S.; Benayoun, B.A.; Marro, S.; Zmoos, A.F.; Cecchini, M.J.; Spacek, D.; Batista, L.F.; O’Brien, M.; et al. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 2015, 16, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Lin, C.P.; Ho, J.J.; He, X.; Okada, N.; Bu, P.; Zhong, Y.; Kim, S.Y.; Bennett, M.J.; Chen, C.; et al. miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat. Cell Biol. 2011, 13, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Schvartzman, J.M.; Thompson, C.B.; Finley, L.W.S. Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 2018, 217, 2247–2259. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Pochampalli, M.; Wang, L.Y.; Zou, J.X.; Li, P.S.; Hsu, S.C.; Wang, B.J.; Huang, S.H.; Yang, P.; Yang, J.C.; et al. KDM8/JMJD5 as a dual coactivator of AR and PKM2 integrates AR/EZH2 network and tumor metabolism in CRPC. Oncogene 2019, 38, 17–32. [Google Scholar] [CrossRef]
- Choi, Y.K.; Park, K.G. Targeting Glutamine Metabolism for Cancer Treatment. Biomol. Ther. 2018, 26, 19–28. [Google Scholar] [CrossRef]
- Choi, S.Y.C.; Ettinger, S.L.; Lin, D.; Xue, H.; Ci, X.; Nabavi, N.; Bell, R.H.; Mo, F.; Gout, P.W.; Fleshner, N.E.; et al. Targeting MCT4 to reduce lactic acid secretion and glycolysis for treatment of neuroendocrine prostate cancer. Cancer Med. 2018, 7, 3385–3392. [Google Scholar] [CrossRef] [PubMed]
- Fiuza-Luces, C.; Garatachea, N.; Berger, N.A.; Lucia, A. Exercise is the real polypill. Physiology 2013, 28, 330–358. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Steensberg, A.; Fischer, C.; Keller, C.; Keller, P.; Plomgaard, P.; Febbraio, M.; Saltin, B. Searching for the exercise factor: Is IL-6 a candidate? J. Muscle Res. Cell Motil. 2003, 24, 113–119. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Akerström, T.C.; Nielsen, A.R.; Fischer, C.P. Role of myokines in exercise and metabolism. J. Appl. Physiol. 2007, 103, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Hursting, S.D.; Contois, J.H.; Strom, S.S.; Yamamura, Y.; Babaian, R.J.; Troncoso, P.; Scardino, P.T.; Wheeler, T.M.; Amos, C.I. Leptin and prostate cancer. Prostate 2001, 46, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.O.; Chun, J.Y.; Nadiminty, N.; Lou, W.; Gao, A.C. Interleukin-6 undergoes transition from growth inhibitor associated with neuroendocrine differentiation to stimulator accompanied by androgen receptor activation during LNCaP prostate cancer cell progression. Prostate 2007, 67, 764–773. [Google Scholar] [CrossRef]
- Morris, J.C.; Ramlogan-Steel, C.A.; Yu, P.; Black, B.A.; Mannan, P.; Allison, J.P.; Waldmann, T.A.; Steel, J.C. Vaccination with tumor cells expressing IL-15 and IL-15Rα inhibits murine breast and prostate cancer. Gene Ther. 2014, 21, 393–401. [Google Scholar] [CrossRef]
- Masjedi, A.; Hajizadeh, F.; Beigi Dargani, F.; Beyzai, B.; Aksoun, M.; Hojjat-Farsangi, M.; Zekiy, A.; Jadidi-Niaragh, F. Oncostatin M: A mysterious cytokine in cancers. Int. Immunopharmacol. 2021, 90, 107158. [Google Scholar] [CrossRef]
- Bozoky, B.; Savchenko, A.; Guven, H.; Ponten, F.; Klein, G.; Szekely, L. Decreased decorin expression in the tumor microenvironment. Cancer Med. 2014, 3, 485–491. [Google Scholar] [CrossRef]
- Liu, J.; Song, N.; Huang, Y.; Chen, Y. Irisin inhibits pancreatic cancer cell growth via the AMPK-mTOR pathway. Sci. Rep. 2018, 8, 15247. [Google Scholar] [CrossRef]
- Catoire, M.; Mensink, M.; Kalkhoven, E.; Schrauwen, P.; Kersten, S. Identification of human exercise-induced myokines using secretome analysis. Physiol. Genom. 2014, 46, 256–267. [Google Scholar] [CrossRef]
- Aoi, W.; Naito, Y.; Takagi, T.; Tanimura, Y.; Takanami, Y.; Kawai, Y.; Sakuma, K.; Hang, L.P.; Mizushima, K.; Hirai, Y.; et al. A novel myokine, secreted protein acidic and rich in cysteine (SPARC), suppresses colon tumorigenesis via regular exercise. Gut 2013, 62, 882–889. [Google Scholar] [CrossRef]
- Kim, J.S.; Galvão, D.A.; Newton, R.U.; Gray, E.; Taaffe, D.R. Exercise-induced myokines and their effect on prostate cancer. Nat. Rev. Urol. 2021, 18, 519–542. [Google Scholar] [CrossRef]
- Hojman, P.; Dethlefsen, C.; Brandt, C.; Hansen, J.; Pedersen, L.; Pedersen, B.K. Exercise-induced muscle-derived cytokines inhibit mammary cancer cell growth. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E504–E510. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K. The Physiology of Optimizing Health with a Focus on Exercise as Medicine. Annu. Rev. Physiol. 2019, 81, 607–627. [Google Scholar] [CrossRef] [PubMed]
- Barfeld, S.J.; Itkonen, H.M.; Urbanucci, A.; Mills, I.G. Androgen-regulated metabolism and biosynthesis in prostate cancer. Endocr.-Relat. Cancer 2014, 21, T57–T66. [Google Scholar] [CrossRef] [PubMed]
- Heemers, H.V.; Verhoeven, G.; Swinnen, J.V. Androgen activation of the sterol regulatory element-binding protein pathway: Current insights. Mol. Endocrinol. 2006, 20, 2265–2277. [Google Scholar] [CrossRef]
- Chan, S.C.; Selth, L.A.; Li, Y.; Nyquist, M.D.; Miao, L.; Bradner, J.E.; Raj, G.V.; Tilley, W.D.; Dehm, S.M. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic Acids Res. 2015, 43, 5880–5897. [Google Scholar] [CrossRef]
- Sharma, N.L.; Massie, C.E.; Ramos-Montoya, A.; Zecchini, V.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.G.; et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 2013, 23, 35–47. [Google Scholar] [CrossRef]
- Swinnen, J.V.; Ulrix, W.; Heyns, W.; Verhoeven, G. Coordinate regulation of lipogenic gene expression by androgens: Evidence for a cascade mechanism involving sterol regulatory element binding proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 12975–12980. [Google Scholar] [CrossRef]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11, eaau5758. [Google Scholar] [CrossRef]
- Tousignant, K.D.; Rockstroh, A.; Taherian Fard, A.; Lehman, M.L.; Wang, C.; McPherson, S.J.; Philp, L.K.; Bartonicek, N.; Dinger, M.E.; Nelson, C.C.; et al. Lipid Uptake Is an Androgen-Enhanced Lipid Supply Pathway Associated with Prostate Cancer Disease Progression and Bone Metastasis. Mol. Cancer Res. MCR 2019, 17, 1166–1179. [Google Scholar] [CrossRef]
- Stoykova, G.E.; Schlaepfer, I.R. Lipid Metabolism and Endocrine Resistance in Prostate Cancer, and New Opportunities for Therapy. Int. J. Mol. Sci. 2019, 20, 2626. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, E.; Khosroushahi, A.Y.; Eftekhari, A.; Farajnia, S.; Babaei, H.; Eghbal, M.A. Novel angiotensin receptor blocker, azilsartan induces oxidative stress and NFkB-mediated apoptosis in hepatocellular carcinoma cell line HepG2. Biomed. Pharmacother. Biomed. Pharmacother. 2018, 99, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, E.; Janas, D.; Eftekhari, A.; Zare, N. Application of carbon nanotubes in sensing/monitoring of pancreas and liver cancer. Chemosphere 2022, 302, 134826. [Google Scholar] [CrossRef] [PubMed]
- Eftekhari, A.; Ahmadian, E.; Salatin, S.; Sharifi, S.; Dizaj, S.M.; Khalilov, R.; Hasanzadeh, M. Current analytical approaches in diagnosis of melanoma. TrAC Trends Anal. Chem. 2019, 116, 122–135. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).