
Lactobacillus johnsonii Attenuates Liver Steatosis and Bile Acid Dysregulation in Parenteral Nutrition-Fed Rats



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Real-Time qPCR
2.3. Western Blot Analysis
2.4. Bile Acid Measurement
2.5. Bacterial DNA Extraction and 16S rDNA Sequencing
2.6. L. johnsonii Whole-Genome Sequencing
2.7. Liver Histology and Biochemical Assay
2.8. Biochemical Assay
2.9. BSH Activity Analysis and Apoptosis Assay
2.10. Statistical Analysis
3. Results
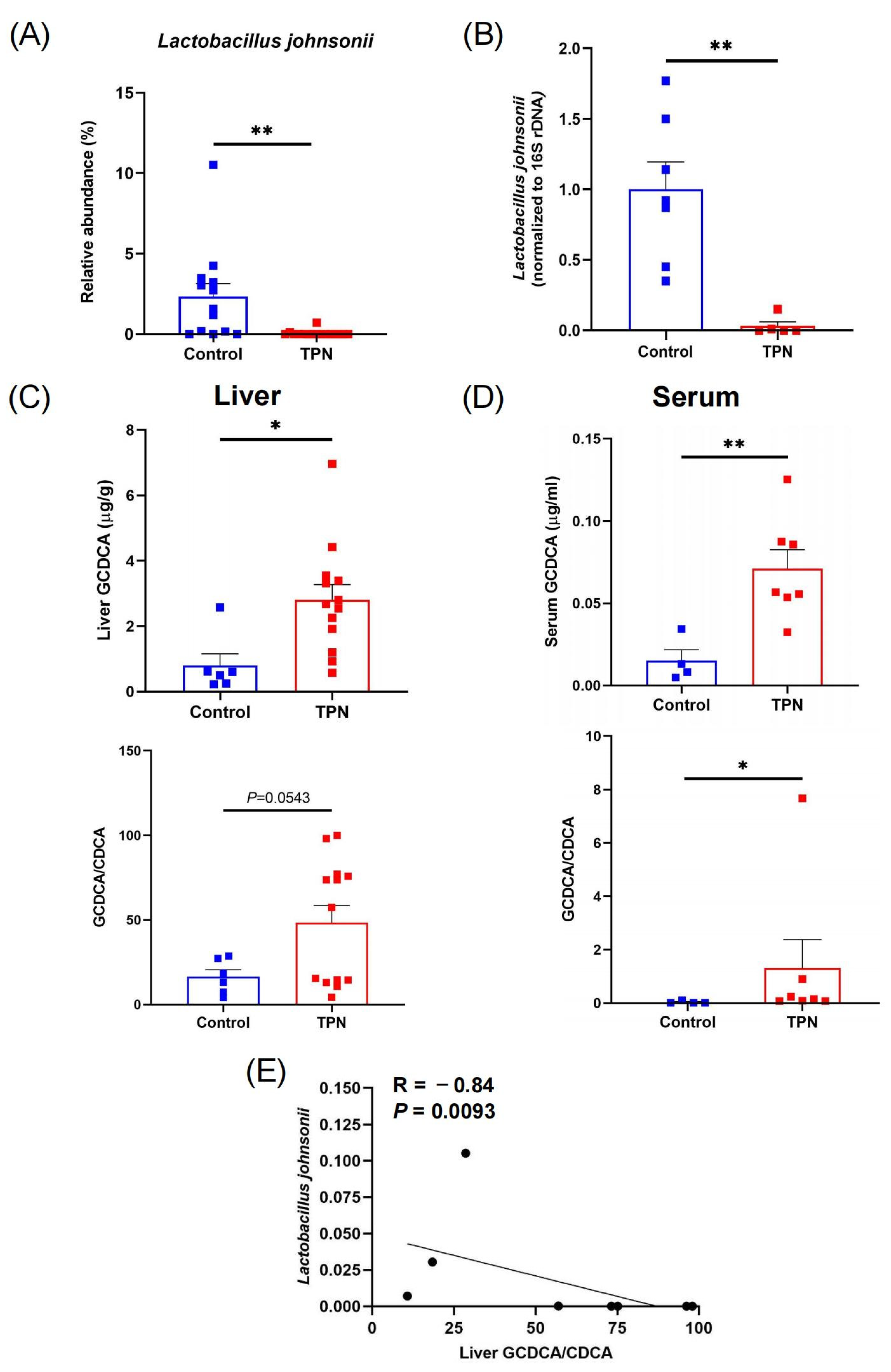
3.1. Reduction in L. johnsonii Is Associated with an Increased GCDCA Level in TPN-Fed Rats
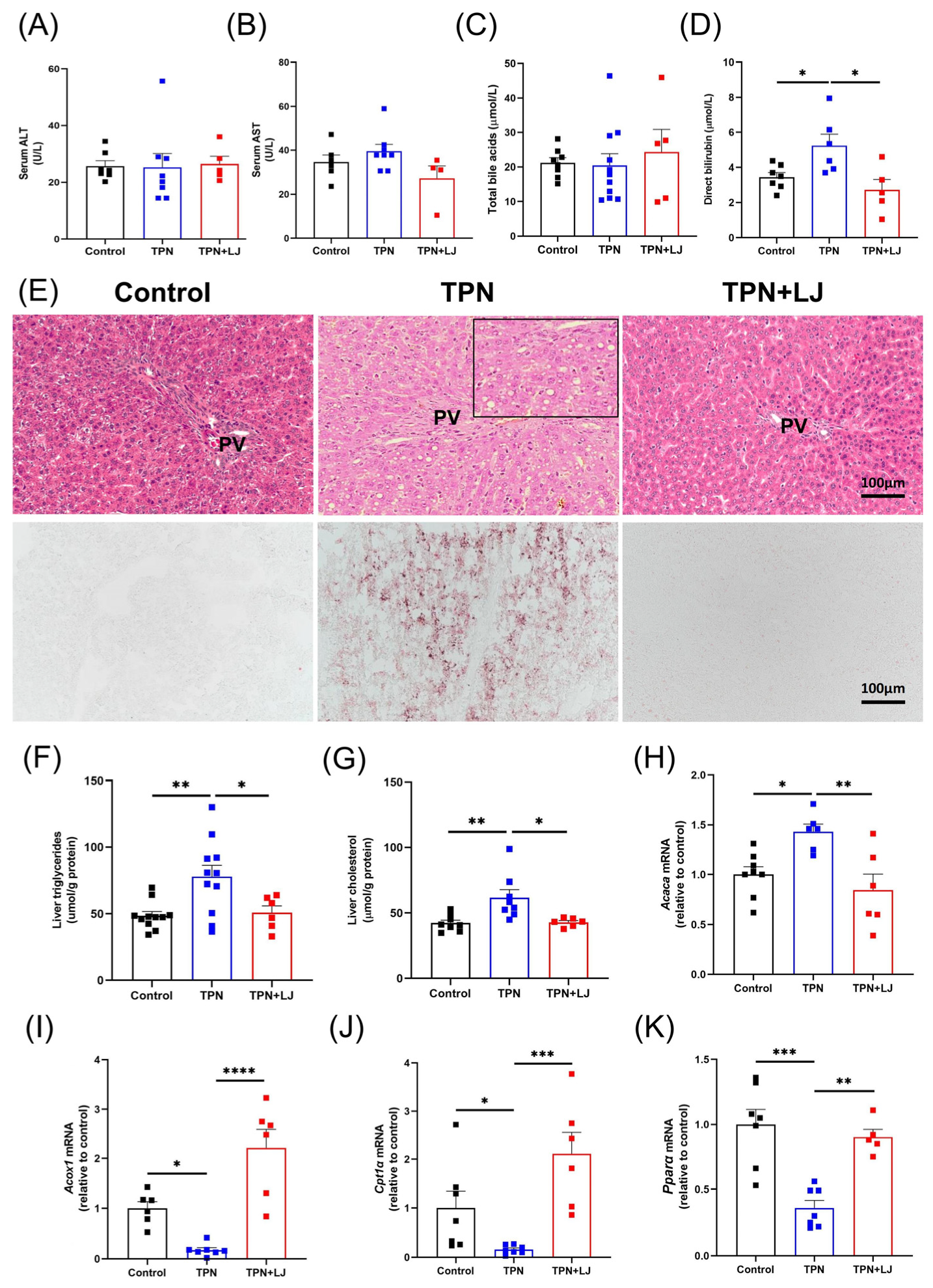
3.2. L. johnsonii Treatment Alleviates Hepatic Steatosis in TPN-Fed Rats
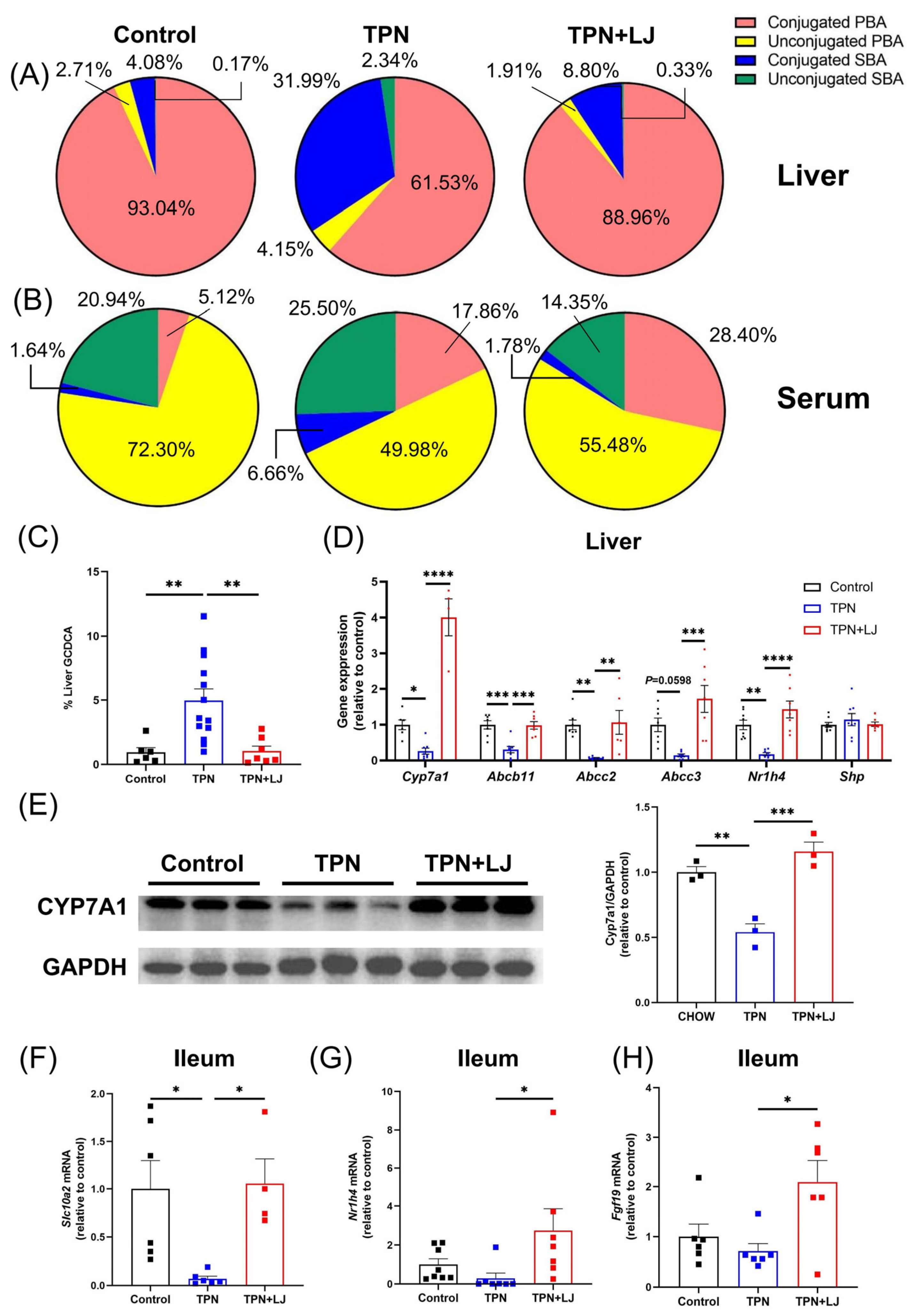
3.3. L. johnsonii Treatment Attenuates TPN-Induced Dysregulation of Bile Acid Metabolism
3.4. L. johnsonii Modulates the Gut Microbiota of TPN-Fed Rats
3.5. L. johnsonii Treatment Reduces Hepatocyte Apoptosis by Deconjugating GCDCA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cederholm, T.; Barazzoni, R.; Austin, P.; Ballmer, P.; Biolo, G.; Bischoff, S.C.; Compher, C.; Correia, I.; Higashiguchi, T.; Holst, M.; et al. ESPEN guidelines on definitions and terminology of clinical nutrition. Clin. Nutr. 2017, 36, 49–64. [Google Scholar] [CrossRef]
- Xu, Z.W.; Li, Y.S. Pathogenesis and treatment of parenteral nutrition-associated liver disease. Hepatobiliary Pancreat. Dis. Int. 2012, 11, 586–593. [Google Scholar] [CrossRef]
- Mutanen, A.; Lohi, J.; Heikkilä, P.; Koivusalo, A.I.; Rintala, R.J.; Pakarinen, M.P. Persistent abnormal liver fibrosis after weaning off parenteral nutrition in pediatric intestinal failure. Hepatology 2013, 58, 729–738. [Google Scholar] [CrossRef]
- Dahlgren, A.F.; Pan, A.; Lam, V.; Gouthro, K.C.; Simpson, P.M.; Salzman, N.H.; Nghiem-Rao, T.H. Longitudinal changes in the gut microbiome of infants on total parenteral nutrition. Pediatr. Res. 2019, 86, 107–114. [Google Scholar] [CrossRef]
- Wang, N.; Wang, J.; Zhang, T.; Huang, L.; Yan, W.; Lu, L.; Jia, J.; Tao, Y.; Cai, W.; Wang, Y. Alterations of gut microbiota and serum bile acids are associated with parenteral nutrition-associated liver disease. J. Pediatr. Surg. 2020, 56, 738–744. [Google Scholar] [CrossRef]
- Cerdó, T.; García-Santos, J.A.; Rodríguez-Pöhnlein, A.; García-Ricobaraza, M.; Nieto-Ruíz, A.; Bermúdez, M.G.; Campoy, C. Impact of Total Parenteral Nutrition on Gut Microbiota in Pediatric Population Suffering Intestinal Disorders. Nutrients 2022, 14, 4691. [Google Scholar] [CrossRef]
- Yang, G.; Hong, E.; Oh, S.; Kim, E. Non-Viable Lactobacillus johnsonii JNU3402 Protects against Diet-Induced Obesity. Foods 2020, 9, 1494. [Google Scholar] [CrossRef]
- Yoon, H.; Lee, Y.; Park, H.; Kang, H.-J.; Ji, Y.; Holzapfel, W.H. Lactobacillus johnsonii BFE6154 Ameliorates Diet-Induced Hypercholesterolemia. Probiotics Antimicrob. Proteins 2021, 15, 451–459. [Google Scholar] [CrossRef]
- Xin, J.; Zeng, D.; Wang, H.; Ni, X.; Yi, D.; Pan, K.; Jing, B. Preventing non-alcoholic fatty liver disease through Lactobacillus johnsonii BS15 by attenuating inflammation and mitochondrial injury and improving gut environment in obese mice. Appl. Microbiol. Biotechnol. 2014, 98, 6817–6829. [Google Scholar] [CrossRef]
- Boutte, H.J., Jr.; Chen, J.; Wylie, T.N.; Wylie, K.M.; Xie, Y.; Geisman, M.; Prabu, A.; Gazit, V.; Tarr, P.I.; Levin, M.S.; et al. Fecal microbiome and bile acid metabolome in adult short bowel syndrome. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 322, G154–G168. [Google Scholar] [CrossRef]
- Koelfat, K.V.K.; Schaap, F.G.; Hodin, C.; Visschers, R.G.; Svavarsson, B.I.; Lenicek, M.; Shiri-Sverdlov, R.; Lenaerts, K.; Damink, S.W.O. Parenteral nutrition dysregulates bile salt homeostasis in a rat model of parenteral nutrition-associated liver disease. Clin. Nutr. 2017, 36, 1403–1410. [Google Scholar] [CrossRef]
- Faubion, W.A.; Guicciardi, M.E.; Miyoshi, H.; Bronk, S.F.; Roberts, P.J.; Svingen, P.A.; Kaufmann, S.H.; Gores, G.J. Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J. Clin. Investig. 1999, 103, 137–145. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, N.; Cheng, S.; Liu, Y.; Chen, S.; Wang, Y.; Cai, W. RNA-sequencing identifies novel transcriptomic signatures in intestinal failure-associated liver disease. J. Pediatr. Surg. 2022, 57, 158–165. [Google Scholar] [CrossRef]
- Xin, F.Z.; Zhao, Z.H.; Liu, X.L.; Pan, Q.; Wang, Z.-X.; Zeng, L.; Zhang, Q.-R.; Ye, L.; Wang, M.-Y.; Zhang, R.-N.; et al. Escherichia fergusonii Promotes Nonobese Nonalcoholic Fatty Liver Disease by Interfering with Host Hepatic Lipid Metabolism through Its Own msRNA 23487. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 827–841. [Google Scholar] [CrossRef]
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511. [Google Scholar] [CrossRef]
- Feng, Z.; Jia, C.; Lin, X.; Hao, H.; Li, S.; Li, F.; Cui, Q.; Chen, Y.; Wu, F.; Xiao, X. The inhibition of enterocyte proliferation by lithocholic acid exacerbates necrotizing enterocolitis through downregulating the Wnt/β-catenin signalling pathway. Cell Prolif. 2022, 55, e13228. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Tang, B.; Tang, L.; Li, S.; Liu, S.; He, J.; Li, P.; Wang, S.; Yang, M.; Zhang, L.; Lei, Y.; et al. Gut microbiota alters host bile acid metabolism to contribute to intrahepatic cholestasis of pregnancy. Nat. Commun. 2023, 14, 1305. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef]
- Patel, T.; Bronk, S.F.; Gores, G.J. Increases of intracellular magnesium promote glycodeoxycholate-induced apoptosis in rat hepatocytes. J. Clin. Investig. 1994, 94, 2183–2192. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, Y.; Zhou, Y.; Xu, Q.; Cheng, S.; Yan, J.; Xiao, Y.; Han, L.; Wang, Y.; Cai, W. Targeted metabolomics unravels altered phenylalanine levels in piglets receiving total parenteral nutrition. FASEB J. 2023, 37, e23014. [Google Scholar] [CrossRef]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef]
- Matsukubo, M.; Yano, K.; Kaji, T.; Sugita, K.; Onishi, S.; Harumatsu, T.; Nagano, A.; Matsui, M.; Murakami, M.; Yamada, K.; et al. The administration of hepatocyte growth factor prevents total parenteral nutrition-induced hepatocellular injury in a rat model. Pediatr. Surg. Int. 2021, 37, 353–361. [Google Scholar] [CrossRef]
- Xiao, Y.T.; Cao, Y.; Zhou, K.J.; Lu, L.-N.; Cai, W. Altered systemic bile acid homeostasis contributes to liver disease in pediatric patients with intestinal failure. Sci. Rep. 2016, 6, 39264. [Google Scholar] [CrossRef]
- Rust, C.; Wild, N.; Bernt, C.; Vennegeerts, T.; Wimmer, R.; Beuers, U. Bile acid-induced apoptosis in hepatocytes is caspase-6-dependent. J. Biol. Chem. 2009, 284, 2908–2916. [Google Scholar] [CrossRef]
- Jang, H.R.; Park, H.J.; Kang, D.; Chung, H.; Nam, M.H.; Lee, Y.; Park, J.-H.; Lee, H.-Y. A protective mechanism of probiotic Lactobacillus against hepatic steatosis via reducing host intestinal fatty acid absorption. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Ritze, Y.; Bárdos, G.; Claus, A.; Ehrmann, V.; Bergheim, I.; Schwiertz, A.; Bischoff, S.C. Lactobacillus rhamnosus GG protects against non-alcoholic fatty liver disease in mice. PLoS ONE 2014, 9, e80169. [Google Scholar] [CrossRef]
- Duerksen, D.R.; Van Aerde, J.E.; Chan, G.; Thomson, A.B.R.; Jewell, L.J.; Clandinin, M.T. Total parenteral nutrition impairs bile flow and alters bile composition in newborn piglet. Dig. Dis. Sci. 1996, 41, 1864–1870. [Google Scholar] [CrossRef]
- Lavoie, J.C.; Chessex, P.; Gauthier, C.; Levy, E.; Alvarez, F.; St-Louis, P.; Rouleau, T. Reduced bile flow associated with parenteral nutrition is independent of oxidant load and parenteral multivitamins. J. Pediatr. Gastroenterol. Nutr. 2005, 41, 108–114. [Google Scholar] [CrossRef]
- Zhan, L.; Yang, I.; Kong, B.; Shen, J.; Gorczyca, L.; Memon, N.; Buckley, B.T.; Guo, G.L.; Pandey, S.; Pierre, J.F.; et al. Dysregulation of bile acid homeostasis in parenteral nutrition mouse model. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G93–G102. [Google Scholar] [CrossRef]
- Zhou, R.; Llorente, C.; Cao, J.; Zaramela, L.S.; Zeng, S.; Gao, B.; Li, S.-Z.; Welch, R.D.; Huang, F.-Q.; Qi, L.-W.; et al. Intestinal α1-2-Fucosylation Contributes to Obesity and Steatohepatitis in Mice. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 293–320. [Google Scholar] [CrossRef]
- Schreuder, T.C.; Marsman, H.A.; Lenicek, M.; van Werven, J.R.; Nederveen, A.J.; Jansen, P.L.M.; Schaap, F.G. The hepatic response to FGF19 is impaired in patients with nonalcoholic fatty liver disease and insulin resistance. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G440–G445. [Google Scholar] [CrossRef]
- Bechmann, L.P.; Kocabayoglu, P.; Sowa, J.P.; Sydor, S.; Best, J.; Schlattjan, M.; Beilfuss, A.; Schmitt, J.; Hannivoort, R.A.; Kilicarslan, A.; et al. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology 2013, 57, 1394–1406. [Google Scholar] [CrossRef]
- Fu, T.; Choi, S.E.; Kim, D.H.; Seok, S.; Suino-Powell, K.M.; Xu, H.E.; Kemper, J.K. Aberrantly elevated microRNA-34a in obesity attenuates hepatic responses to FGF19 by targeting a membrane coreceptor β-Klotho. Proc. Natl. Acad. Sci. USA 2012, 109, 16137–16142. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, Y.; Xiao, Y.; Wang, Y.; Yan, J.; Schnabl, B.; Cai, W. Role of the Gut Microbiota in Parenteral Nutrition-Associated Liver Disease: From Current Knowledge to Future Opportunities. J. Nutr. 2022, 152, 377–385. [Google Scholar] [CrossRef]
- Demehri, F.R.; Barrett, M.; Ralls, M.W.; Miyasaka, E.A.; Feng, Y.; Teitelbaum, D.H. Intestinal epithelial cell apoptosis and loss of barrier function in the setting of altered microbiota with enteral nutrient deprivation. Front. Cell. Infect. Microbiol. 2013, 3, 105. [Google Scholar] [CrossRef]
- Cao, P.; Chen, Q.; Shi, C.; Wang, L.; Gong, Z. Fusobacterium nucleatum promotes the development of acute liver failure by inhibiting the NAD(+) salvage metabolic pathway. Gut Pathog. 2022, 14, 29. [Google Scholar] [CrossRef]
- Esmat, G.; El-Bendary, M.; Zakarya, S.; Ela, M.A.; Zalata, K. Role of Helicobacter pylori in patients with HCV-related chronic hepatitis and cirrhosis with or without hepatocellular carcinoma: Possible association with disease progression. J. Viral Hepat. 2012, 19, 473–479. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Papatheodorou, A.; Patsiaoura, K.; Katsiki, E.; Zafeiriadou, E.; Zavos, C.; Anastasiadou, K.; Terpos, E. Helicobacter pylori infection in patients with nonalcoholic fatty liver disease. Metabolism 2013, 62, 121–126. [Google Scholar] [CrossRef]
- Rocha, M.; Avenaud, P.; Ménard, A.; Le Bail, B.; Balabaud, C.; Bioulac-Sage, P.; de Magalhães Queiroz, D.M.; Mégraud, F. Association of Helicobacter species with hepatitis C cirrhosis with or without hepatocellular carcinoma. Gut 2005, 54, 396–401. [Google Scholar] [CrossRef]
- Muhtaroglu, A.; Sengul, I.; Sengul, D.; Kesicioglu, T.; Seker, D.; Aydin, M.; Dulger, A.C. Does enteral nutrition through a percutaneous endoscopic gastrostomy, attenuate Helicobacter pylori colonization?: Is it worth mentioning? Rev. Assoc. Med. Bras. (1992) 2023, 69, e20221733. [Google Scholar] [CrossRef]
- Buchman, A.L.; Moukarzel, A.A.; Bhuta, S.; Belle, M.; Ament, M.E.; Eckhert, C.D.; Hollander, D.; Gornbeln, J.; Kopple, J.D.; Vijayaroghavan, S.R. Parenteral nutrition is associated with intestinal morphologic and functional changes in humans. JPEN J. Parenter. Enteral. Nutr. 1995, 19, 453–460. [Google Scholar] [CrossRef]
- Burrin, D.G.; Stoll, B.; Jiang, R.; Petersen, Y.; Elnif, J.; Buddington, R.K.; Schmidt, M.; Holst, J.J.; Hartmann, B.; Sangild, P.T. GLP-2 stimulates intestinal growth in premature TPN-fed pigs by suppressing proteolysis and apoptosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G1249-56. [Google Scholar] [CrossRef]
- Yang, H.; Fan, Y.; Teitelbaum, D.H. Intraepithelial lymphocyte-derived interferon-gamma evokes enterocyte apoptosis with parenteral nutrition in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G629–G637. [Google Scholar] [CrossRef]
- Allain, T.; Chaouch, S.; Thomas, M.; Vallée, I.; Buret, A.G.; Langella, P.; Grellier, P.; Polack, B.; Bermúdez-Humarán, L.G.; Florent, I. Bile-Salt-Hydrolases from the Probiotic Strain Lactobacillus johnsonii La1 Mediate Anti-giardial Activity In Vitro and In Vivo. Front. Microbiol. 2017, 8, 2707. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Sequence | Reverse Sequence |
|---|---|---|
| Abcb11 | CTGCCAAGGATGCTAATGCA | CGATGGCTACCCTTTGCTTCT |
| Abcc2 | TCGAGAGAGGCTGACCATCA | TCTGCCCTATGCTCAGGTTG |
| Abcc3 | GCCTTACAGGTGACCTTGAGTT | CGGTACCGCACCGAATAGTT |
| Acaca | CCCAGAGATGTTTCGGCAGTCAC | GTCAGGATGTCGGAAGGCAAAGG |
| Acox1 | AGTCTGAAATCAAGCAAAGC | CATTAATTCGAAGGTAGGTCTC |
| Cpt1α | TAGGACAGGCAGAAAATTGC | CAGTAGGAGCCGATTCAAAA |
| Cyp7a1 | TGCCTTCTGTTACCGAGTGATGT | ACCGGCAGGTCATTCAGTTGCACT |
| Fgf19 | ATACGGGCTGATTCGCTACT | GCTGGTCCGTGGATTCAAAG |
| Nr0b2 | CGATCCTCTTCAACCCAGATG | AGGGCTCCAAGACTTCACACA |
| Nr1h4 | GAAACTGAACATCGGGGTTAT | CGGCGGAGATTTTCAATAAG |
| Ntcp | CAAACCTCAGAAGGACCAAACA | GTAGGAGGATTATTCCCGTTGTG |
| Pparα | AATGCAATCCGTTTTGGAAG | TTGGCCAGAGATTTGAGGTC |
| Slc10a2 | TGGTGTAGACGAAGAGGCAA | GCCTATTGGATAGATGGCGA |
| 18S | ACGGAAGGGCACCACCAGGA | CACCACCACCCACGGAATCG |
| 16S | GTGSTGCAYGGYTGTCGTCA | ACGTCRTCCMCACCTTCCTC |
| L. johnsonii | AGAGAGAAACTCAACTTGAAATA | CCTTCATTAACCTTAACAGTTAA |
| Control vs. TPN | TPN vs. TPN + LJ | |
|---|---|---|
| Liver | ||
| % unconjugated PBA/TBA | 0.4071 | 0.1041 |
| % conjugated PBA/TBA | <0.0001 | <0.0001 |
| % unconjugated SBA/TBA | 0.0369 | 0.0429 |
| % conjugated SBA/TBA | <0.0001 | <0.0001 |
| Serum | ||
| % unconjugated PBA/TBA | 0.0374 | 0.7035 |
| % conjugated PBA/TBA | 0.6261 | 0.6387 |
| % unconjugated SBA/TBA | 0.4235 | 0.1033 |
| % conjugated SBA/TBA | 0.0227 | 0.0478 |
| Gene ID | Identity | E Value | KO ID | KO Definition |
|---|---|---|---|---|
| LJ4_GM000177 | 99.1 | 9.40 × 10−185 | K01442 | choloylglycine hydrolase |
| LJ4_GM000897 | 100 | 3.10 × 10−191 | K01442 | choloylglycine hydrolase |
| LJ4_GM001072 | 99.4 | 1.20 × 10−187 | K01442 | choloylglycine hydrolase |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Zhou, Y.; Cheng, S.; Zhao, Y.; Yan, J.; Wang, Y.; Cai, W.; Jiang, L. Lactobacillus johnsonii Attenuates Liver Steatosis and Bile Acid Dysregulation in Parenteral Nutrition-Fed Rats. Metabolites 2023, 13, 1043. https://doi.org/10.3390/metabo13101043
Xu J, Zhou Y, Cheng S, Zhao Y, Yan J, Wang Y, Cai W, Jiang L. Lactobacillus johnsonii Attenuates Liver Steatosis and Bile Acid Dysregulation in Parenteral Nutrition-Fed Rats. Metabolites. 2023; 13(10):1043. https://doi.org/10.3390/metabo13101043
Chicago/Turabian StyleXu, Juan, Yongchang Zhou, Siyang Cheng, Yuling Zhao, Junkai Yan, Ying Wang, Wei Cai, and Lu Jiang. 2023. "Lactobacillus johnsonii Attenuates Liver Steatosis and Bile Acid Dysregulation in Parenteral Nutrition-Fed Rats" Metabolites 13, no. 10: 1043. https://doi.org/10.3390/metabo13101043
APA StyleXu, J., Zhou, Y., Cheng, S., Zhao, Y., Yan, J., Wang, Y., Cai, W., & Jiang, L. (2023). Lactobacillus johnsonii Attenuates Liver Steatosis and Bile Acid Dysregulation in Parenteral Nutrition-Fed Rats. Metabolites, 13(10), 1043. https://doi.org/10.3390/metabo13101043