

The Combination of Bioinformatics Analysis and Untargeted Metabolomics Reveals Potential Biomarkers and Key Metabolic Pathways in Asthma



Abstract

1. Introduction
2. Materials and Methods
2.1. Data Download
2.2. Identify Molecular Subtypes Using NMF Algorithm
2.3. The screening of Differentially Expressed Genes (DEGs)
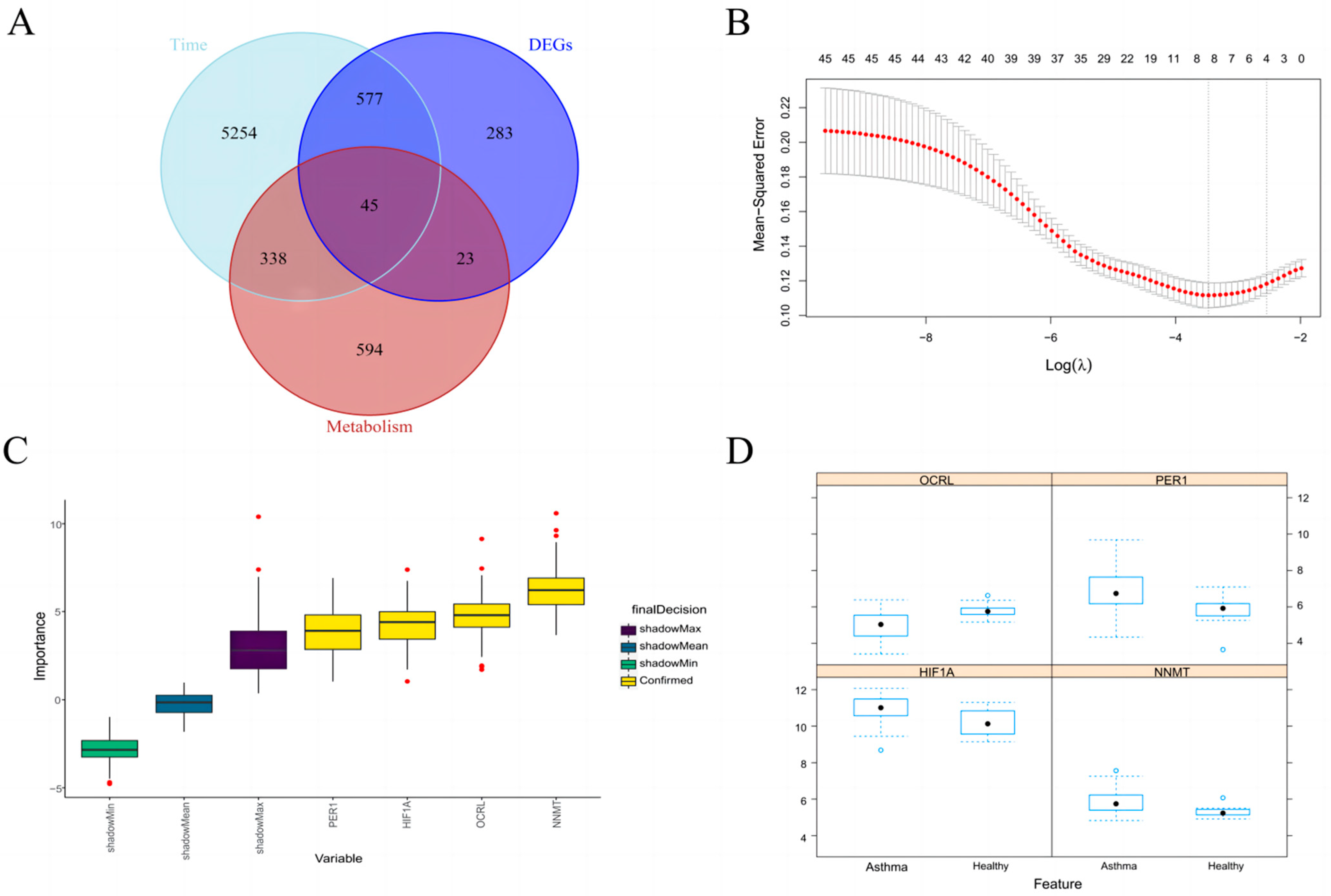
2.4. Time Series Analysis and the Intersection of Metabolic Genes and DEGs
2.5. LASSO Regression and Correlation Analysis
2.6. Random Forest
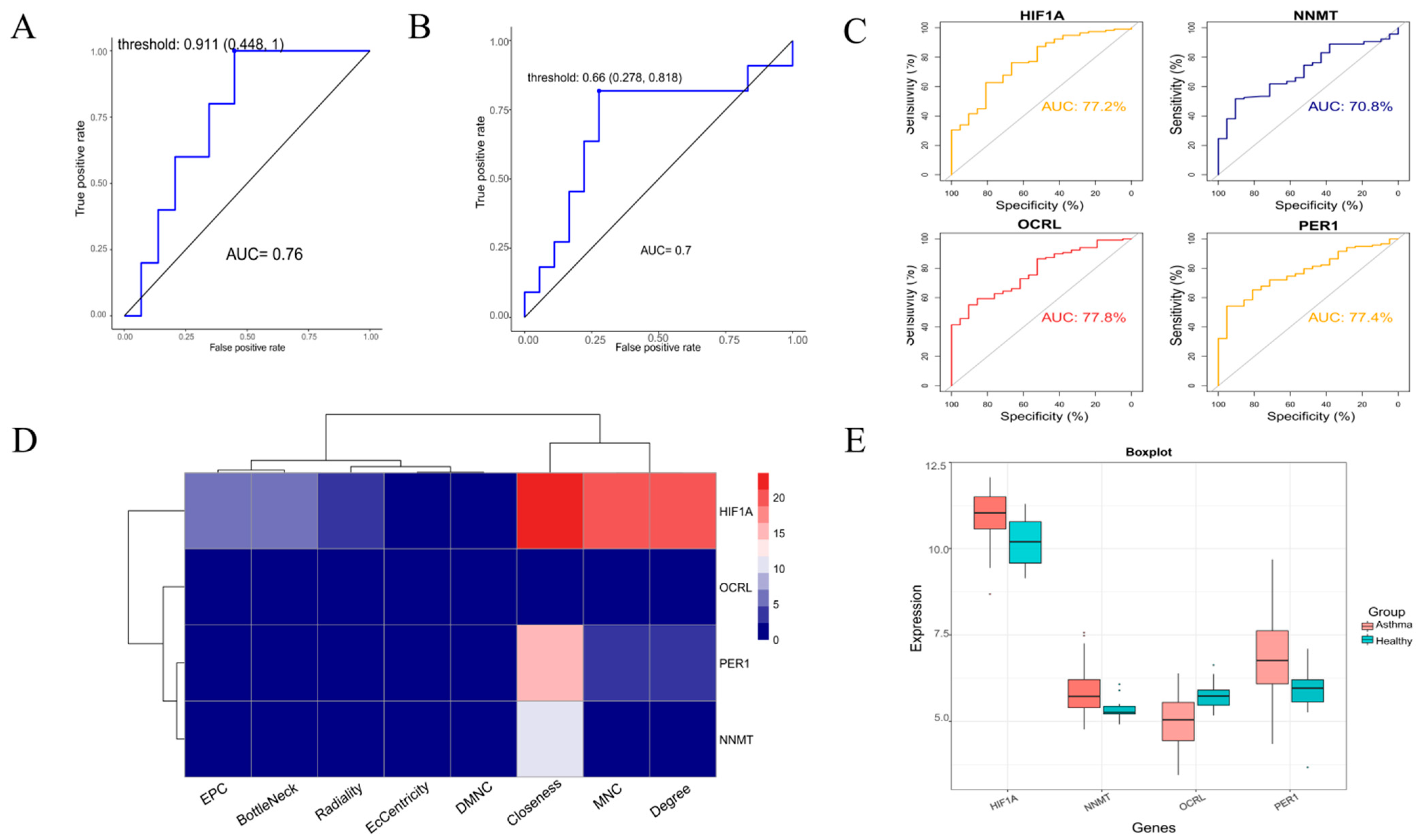
2.7. ROC Analysis of Risk Models
2.8. Gene Expression and Hub Gene Screening
2.9. Functional Enrichment Analysis
2.10. Identification of Infiltrating Immune Cells in Asthma Samples
2.11. Reagents and Antibodies
2.12. Cell Culture and Treatment
2.13. Western Blotting
2.14. Immunofluorescence
2.15. Untargeted Metabolomics Sample Collection and Preparation
2.16. LC-MS/MS Analysis
2.17. Untargeted Metabolomics Data Processing
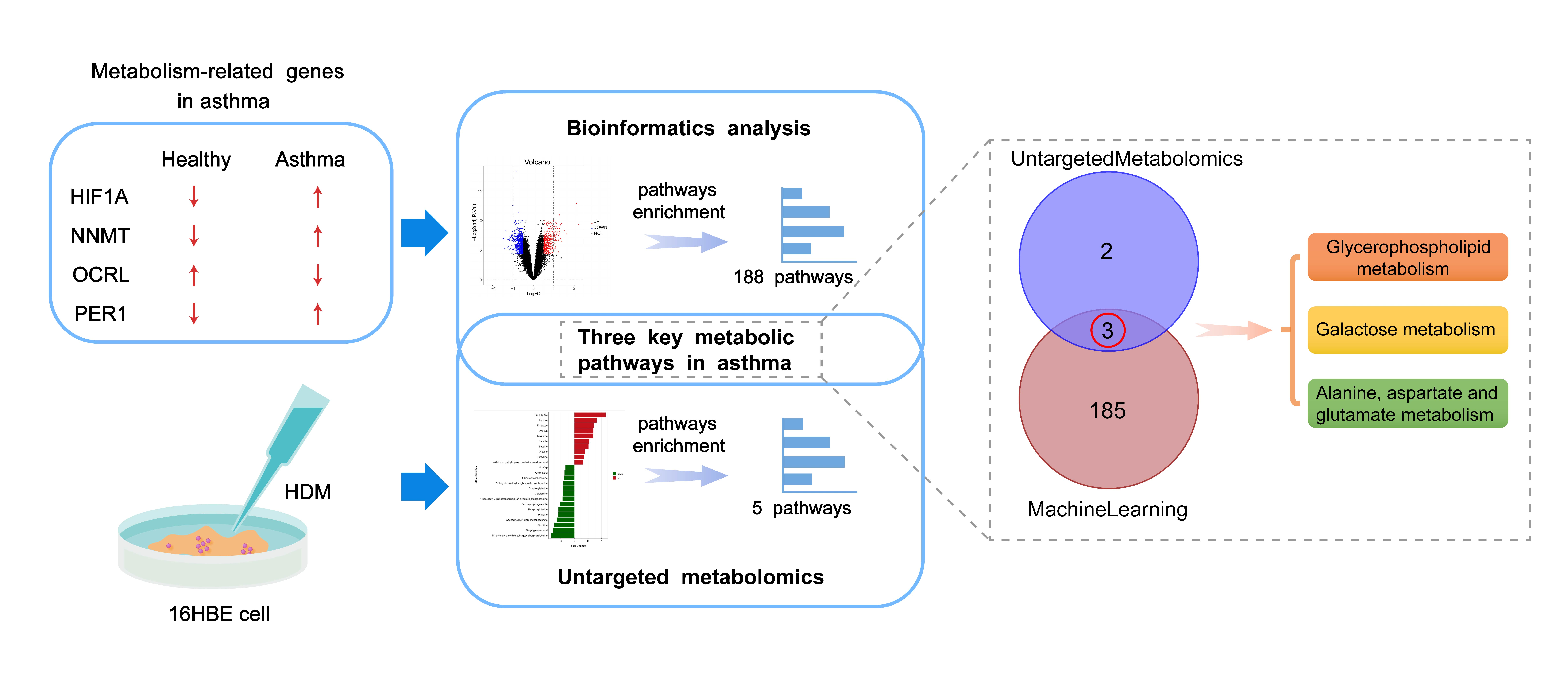
2.18. Combined Analysis of Bioinformatics Analysis and Untargeted Metabolomics
2.19. Statistical Analysis
3. Results
3.1. Molecular Typing and Identification of DEGs
3.2. Time Series Analysis and Take Intersection
3.3. Identification of Key Metabolic Gene Associated with Asthma
3.4. Random Forest further Screen Hub Genes
3.5. Model Validation and Expression Level Analysis of Hub Genes
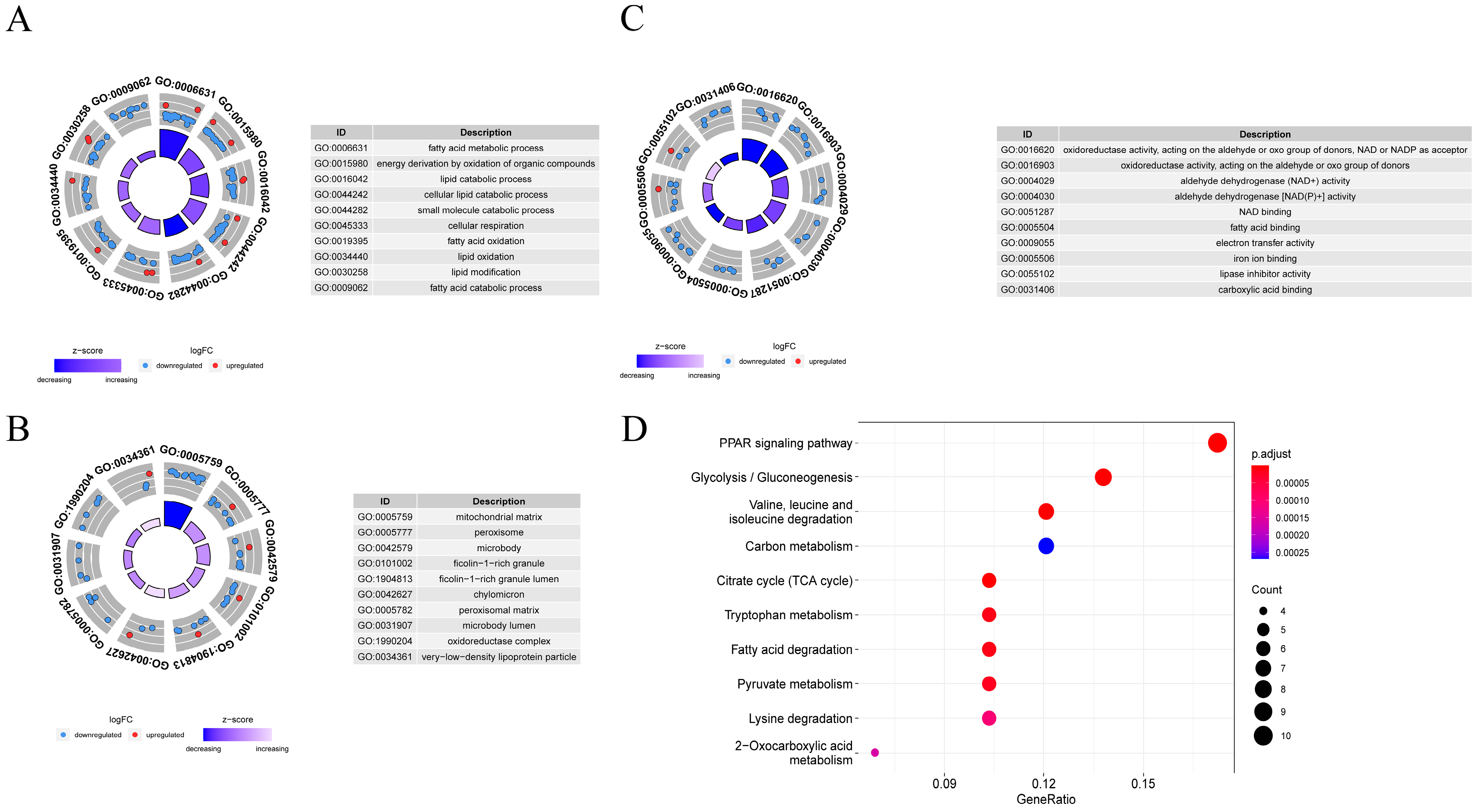
3.6. Function and Pathway Enrichment Analysis
3.7. HIF1A-Enriched Pathways and Immune Infiltrates Analysis
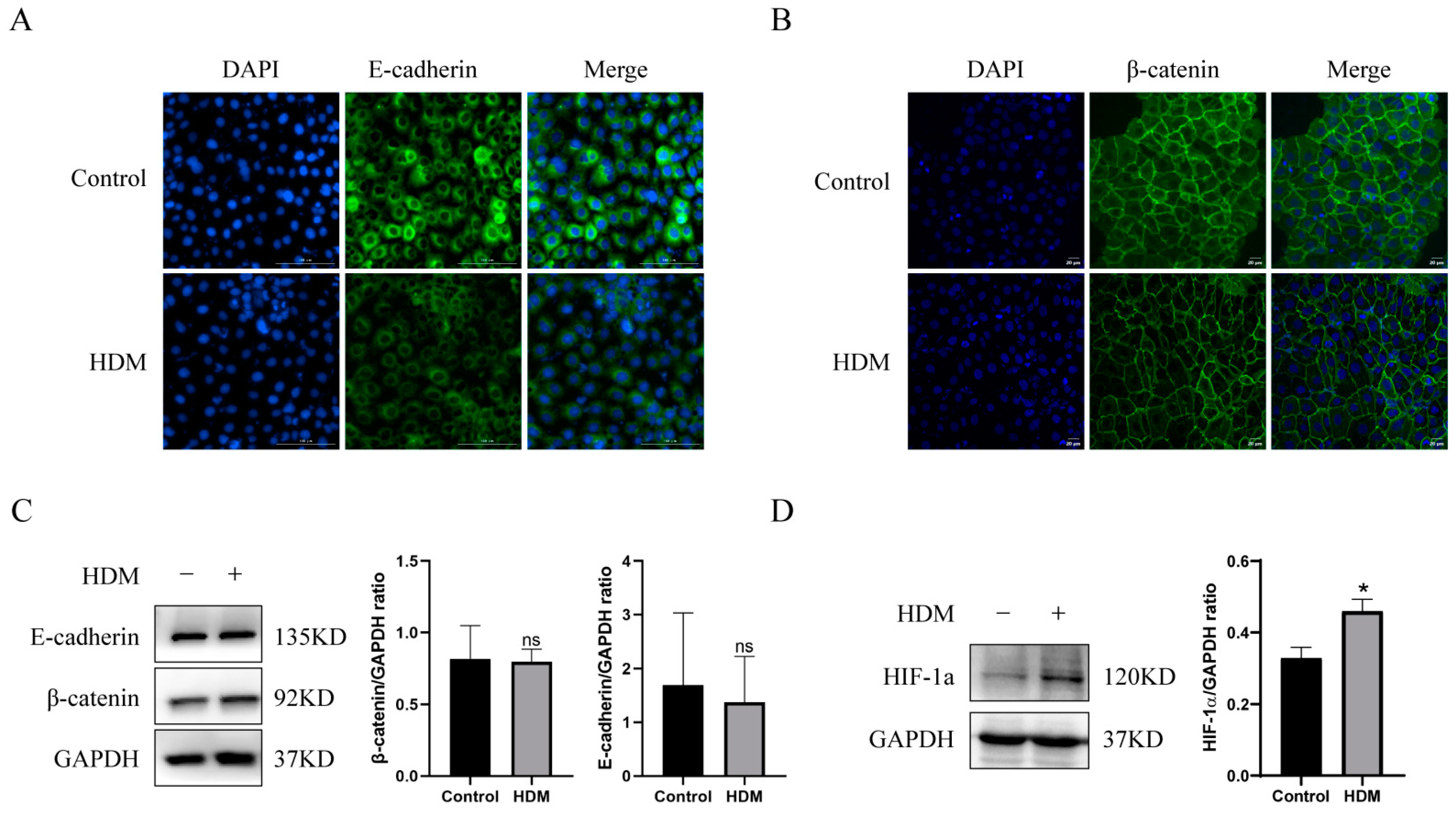
3.8. HDM Induced Delocalization of E-cadherin and β-catenin and Promoted the Expression of HIF-1a in 16HBE Cells
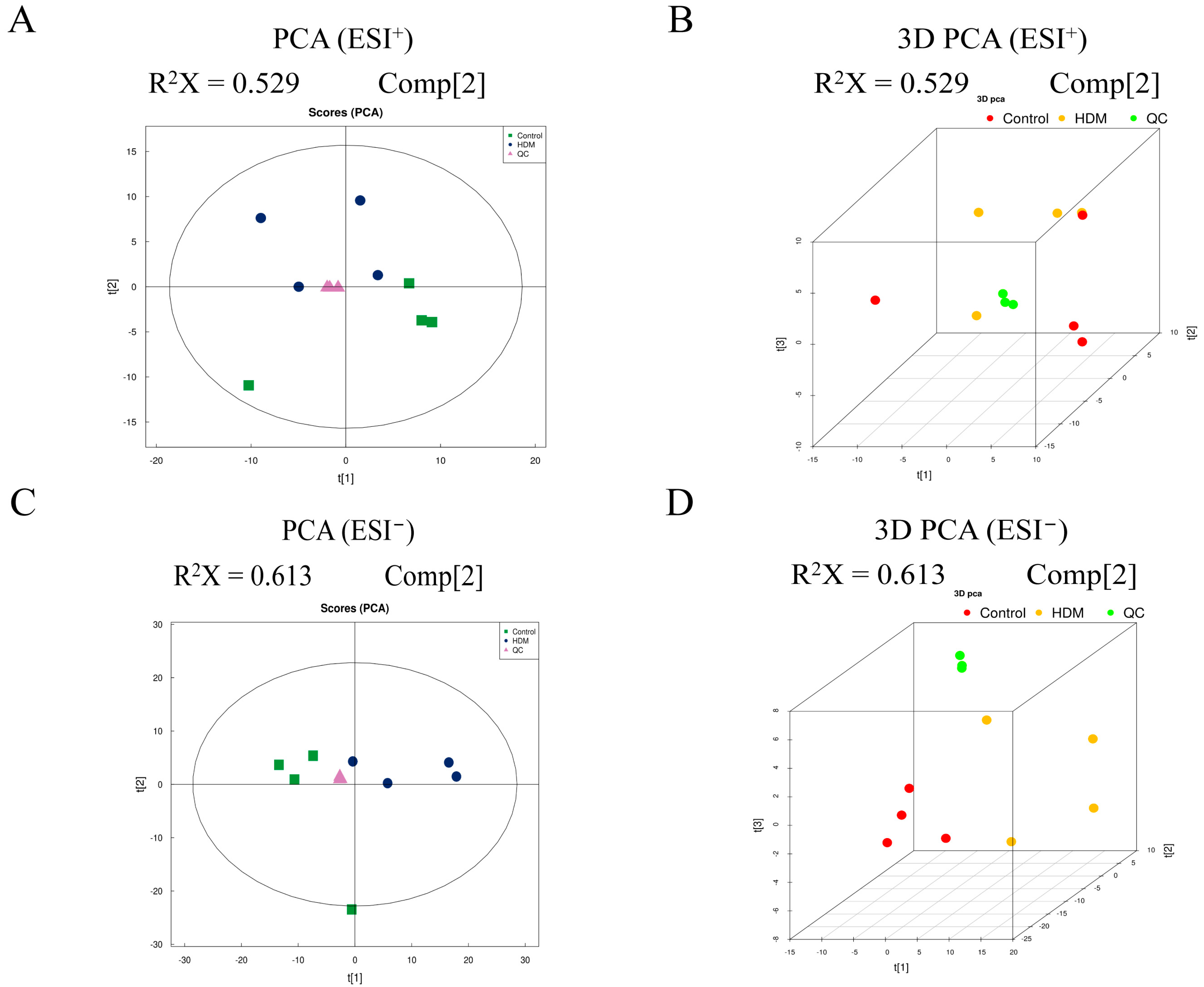
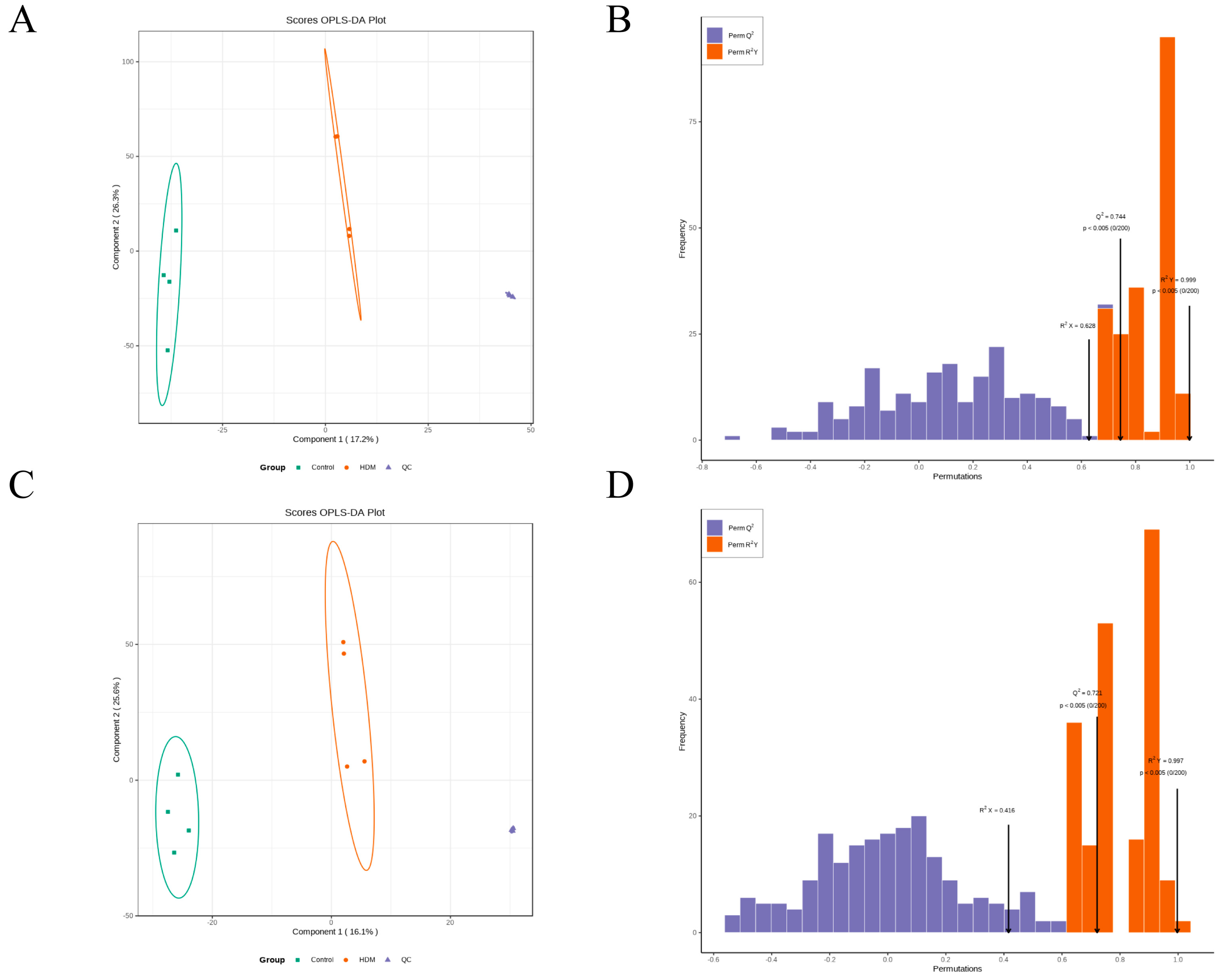
3.9. Multivariate Analysis of Metabolomic Data
3.10. Identification of the Differential Metabolites
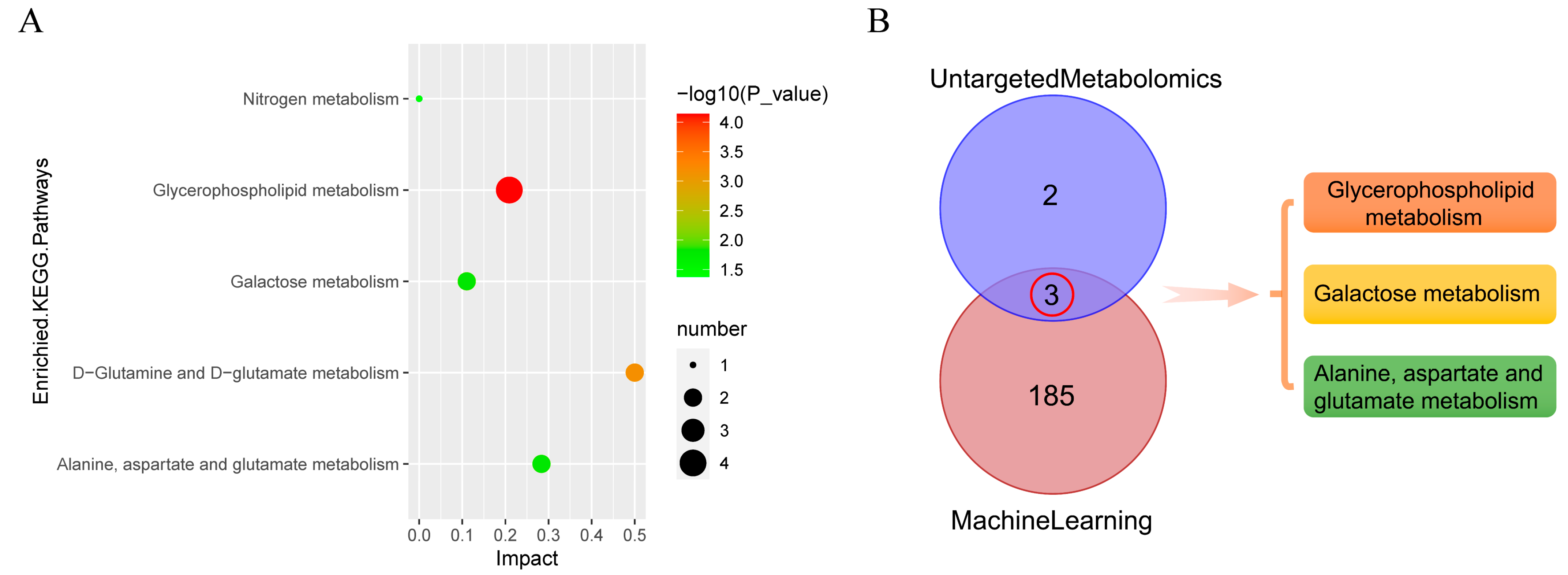
3.11. Metabolic Pathway Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Papi, A.; Brightling, C.; Pedersen, S.E.; Reddel, H.K. Asthma. Lancet 2018, 391, 783–800. [Google Scholar] [CrossRef]
- Taqi, A.; Rowlands, G.; Rathbone, A.P. A systematic review and thematic synthesis to identify factors that influence pharmacists’ involvement in asthma care services: An identity crisis. Explor. Res. Clin. Soc. Pharm. 2021, 3, 100051. [Google Scholar] [CrossRef]
- Fahy, J.V. Type 2 inflammation in asthma--present in most, absent in many. Nat. Rev. Immunol. 2015, 15, 57–65. [Google Scholar] [CrossRef]
- Brannan, J.D.; Lougheed, M.D. Airway hyperresponsiveness in asthma: Mechanisms, clinical significance, and treatment. Front Physiol 2012, 3, 460. [Google Scholar] [CrossRef]
- Huang, K.; Yang, T.; Xu, J.; Yang, L.; Zhao, J.; Zhang, X.; Bai, C.; Kang, J.; Ran, P.; Shen, H.; et al. Prevalence, risk factors, and management of asthma in China: A national cross-sectional study. Lancet 2019, 394, 407–418. [Google Scholar] [CrossRef]
- Newton, R.; Leigh, R.; Giembycz, M.A. Pharmacological strategies for improving the efficacy and therapeutic ratio of glucocorticoids in inflammatory lung diseases. Pharmacol. Ther 2010, 125, 286–327. [Google Scholar]
- Bateman, E.D.; Hurd, S.S.; Barnes, P.J.; Bousquet, J.; Drazen, J.M.; FitzGerald, J.M.; Gibson, P.; Ohta, K.; O’Byrne, P.; Pedersen, S.E.; et al. Global strategy for asthma management and prevention: GINA executive summary. Eur. Respir. J. 2008, 31, 143–178. [Google Scholar] [CrossRef] [PubMed]
- German, J.B.; Hammock, B.D.; Watkins, S.M. Metabolomics: Building on a century of biochemistry to guide human health. Metabolomics 2005, 1, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic. Acids. Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef] [PubMed]
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Untargeted Metabolomics Strategies-Challenges and Emerging Directions. J. Am. Soc. Mass. Spectrom. 2016, 27, 1897–1905. [Google Scholar] [CrossRef]
- Rinschen, M.M.; Ivanisevic, J.; Giera, M.; Siuzdak, G. Identification of bioactive metabolites using activity metabolomics. Nat. Rev. Mol. Cell. Biol. 2019, 20, 353–367. [Google Scholar] [CrossRef]
- Pacchiarotta, T.; Deelder, A.M.; Mayboroda, O.A. Metabolomic investigations of human infections. Bioanalysis 2012, 4, 919–925. [Google Scholar] [CrossRef]
- Gieger, C.; Geistlinger, L.; Altmaier, E.; Hrabé, D.A.M.; Kronenberg, F.; Meitinger, T.; Mewes, H.W.; Wichmann, H.E.; Weinberger, K.M.; Adamski, J.; et al. Genetics meets metabolomics: A genome-wide association study of metabolite profiles in human serum. PLoS Genet. 2008, 4, e1000282. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic. Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Safran, M.; Dalah, I.; Alexander, J.; Rosen, N.; Iny, S.T.; Shmoish, M.; Nativ, N.; Bahir, I.; Doniger, T.; Krug, H.; et al. GeneCards Version 3: The human gene integrator. Database (Oxford) 2010, 2010, baq020. [Google Scholar] [CrossRef]
- Georas, S.N.; Rezaee, F. Epithelial barrier function: At the front line of asthma immunology and allergic airway inflammation. J. Allergy Clin. Immunol. 2014, 134, 509–520. [Google Scholar] [CrossRef]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2022, 23, 40–55. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, K.S.; Park, H.S.; Park, S.J.; Min, K.H.; Moon, H.; Puri, K.D.; Lee, Y.C. HIF-1α inhibition ameliorates an allergic airway disease via VEGF suppression in bronchial epithelium. Eur. J. Immunol. 2010, 40, 2858–2869. [Google Scholar] [CrossRef]
- Huerta-Yepez, S.; Baay-Guzman, G.J.; Bebenek, I.G.; Hernandez-Pando, R.; Vega, M.I.; Chi, L.; Riedl, M.; Diaz-Sanchez, D.; Kleerup, E.; Tashkin, D.P.; et al. Hypoxia inducible factor promotes murine allergic airway inflammation and is increased in asthma and rhinitis. Allergy 2011, 66, 909–918. [Google Scholar] [CrossRef]
- Dewitz, C.; McEachern, E.; Shin, S.; Akong, K.; Nagle, D.G.; Broide, D.H.; Akuthota, P.; Crotty, A.L. Hypoxia-inducible factor-1α inhibition modulates airway hyperresponsiveness and nitric oxide levels in a BALB/c mouse model of asthma. Clin. Immunol. 2017, 176, 94–99. [Google Scholar] [CrossRef]
- Guo, J.; Lu, W.; Shimoda, L.A.; Semenza, G.L.; Georas, S.N. Enhanced interferon-gamma gene expression in T Cells and reduced ovalbumin-dependent lung eosinophilia in hypoxia-inducible factor-1-alpha-deficient mice. Int. Arch. Allergy Immunol. 2009, 149, 98–102. [Google Scholar] [CrossRef]
- Sun, L.; Fu, J.; Lin, S.H.; Sun, J.L.; Xia, L.; Lin, C.H.; Liu, L.; Zhang, C.; Yang, L.; Xue, P.; et al. Particulate matter of 2.5 μm or less in diameter disturbs the balance of T(H)17/regulatory T cells by targeting glutamate oxaloacetate transaminase 1 and hypoxia-inducible factor 1α in an asthma model. J. Allergy Clin. Immunol. 2020, 145, 402–414. [Google Scholar] [CrossRef]
- Jung, J.; Kim, S.H.; Lee, H.S.; Choi, G.S.; Jung, Y.S.; Ryu, D.H.; Park, H.S.; Hwang, G.S. Serum metabolomics reveals pathways and biomarkers associated with asthma pathogenesis. Clin. Exp. Allergy 2013, 43, 425–433. [Google Scholar] [CrossRef]
- Horgusluoglu, E.; Neff, R.; Song, W.M.; Wang, M.; Wang, Q.; Arnold, M.; Krumsiek, J.; Galindo-Prieto, B.; Ming, C.; Nho, K.; et al. Integrative metabolomics-genomics approach reveals key metabolic pathways and regulators of Alzheimer’s disease. Alzheimers Dement. 2022, 18, 1260–1278. [Google Scholar] [CrossRef]
- Farraia, M.; Cavaleiro, R.J.; Paciência, I.; Castro, M.F.; Delgado, L.; Laerte, B.J.; Moreira, A. Metabolic interactions in asthma. Eur. Ann. Allergy Clin. Immunol. 2019, 51, 196–205. [Google Scholar] [CrossRef]
- Fitzpatrick, A.M.; Park, Y.; Brown, L.A.; Jones, D.P. Children with severe asthma have unique oxidative stress-associated metabolomic profiles. J. Allergy Clin. Immunol. 2014, 133, 258–261. [Google Scholar] [CrossRef]
- Michaeloudes, C.; Abubakar-Waziri, H.; Lakhdar, R.; Raby, K.; Dixey, P.; Adcock, I.M.; Mumby, S.; Bhavsar, P.K.; Chung, K.F. Molecular mechanisms of oxidative stress in asthma. Mol. Aspects Med. 2022, 85, 101026. [Google Scholar] [CrossRef]
- Lumia, M.; Luukkainen, P.; Takkinen, H.M.; Kaila, M.; Nwaru, B.I.; Nevalainen, J.; Salminen, I.; Uusitalo, L.; Niinistö, S.; Tuokkola, J.; et al. Cow’s milk allergy and the association between fatty acids and childhood asthma risk. J. Allergy Clin. Immunol. 2014, 134, 488–490. [Google Scholar] [CrossRef]
- Lee-Sarwar, K.; Kelly, R.S.; Lasky-Su, J.; Kachroo, P.; Zeiger, R.S.; O’Connor, G.T.; Sandel, M.T.; Bacharier, L.B.; Beigelman, A.; Laranjo, N.; et al. Dietary and Plasma Polyunsaturated Fatty Acids Are Inversely Associated with Asthma and Atopy in Early Childhood. J. Allergy Clin. Immunol. Pract. 2019, 7, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, J.; Ekström, S.; Kull, I.; Håkansson, N.; Nilsson, S.; Wickman, M.; Melén, E.; Risérus, U.; Bergström, A. Polyunsaturated fatty acids in plasma at 8 years and subsequent allergic disease. J. Allergy Clin. Immunol. 2018, 142, 510–516. [Google Scholar] [CrossRef]
- Hirota, J.A.; Knight, D.A. Human airway epithelial cell innate immunity: Relevance to asthma. Curr. Opin. Immunol. 2012, 24, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Puddicombe, S.M.; Field, S.; Haywood, J.; Broughton-Head, V.; Puxeddu, I.; Haitchi, H.M.; Vernon-Wilson, E.; Sammut, D.; Bedke, N.; et al. Defective epithelial barrier function in asthma. J. Allergy Clin. Immunol. 2011, 128, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, P.K.; Wardlaw, A.J.; Nelson, F.C.; Collins, J.V.; Kay, A.B. Bronchial biopsies in asthma. An ultrastructural, quantitative study and correlation with hyperreactivity. Am. Rev. Respir. Dis. 1989, 140, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Heijink, I.H.; Kies, P.M.; Kauffman, H.F.; Postma, D.S.; van Oosterhout, A.J.; Vellenga, E. Down-regulation of E-cadherin in human bronchial epithelial cells leads to epidermal growth factor receptor-dependent Th2 cell-promoting activity. J. Immunol. 2007, 178, 7678–7685. [Google Scholar] [CrossRef] [PubMed]
- Leino, M.S.; Loxham, M.; Blume, C.; Swindle, E.J.; Jayasekera, N.P.; Dennison, P.W.; Shamji, B.W.; Edwards, M.J.; Holgate, S.T.; Howarth, P.H.; et al. Barrier disrupting effects of alternaria alternata extract on bronchial epithelium from asthmatic donors. PLoS ONE 2013, 8, e71278. [Google Scholar] [CrossRef]
- Post, S.; Nawijn, M.C.; Jonker, M.R.; Kliphuis, N.; van den Berge, M.; van Oosterhout, A.J.; Heijink, I.H. House dust mite-induced calcium signaling instigates epithelial barrier dysfunction and CCL20 production. Allergy 2013, 68, 1117–1125. [Google Scholar] [CrossRef]
- Shimizu, T. Lipid mediators in health and disease: Enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 123–150. [Google Scholar] [CrossRef]
- Wang, S.; Tang, K.; Lu, Y.; Tian, Z.; Huang, Z.; Wang, M.; Zhao, J.; Xie, J. Revealing the role of glycerophospholipid metabolism in asthma through plasma lipidomics. Clin. Chim. Acta 2021, 513, 34–42. [Google Scholar] [CrossRef]
- Baritussio, A. Lung surfactant, asthma, and allergens: A story in evolution. Am. J. Respir. Crit. Care Med. 2004, 169, 550–551. [Google Scholar] [CrossRef]
- Guo, Z.; Vikbjerg, A.F.; Xu, X. Enzymatic modification of phospholipids for functional applications and human nutrition. Biotechnol. Adv. 2005, 23, 203–259. [Google Scholar] [CrossRef]
- Jiang, L.; Diaz, P.T.; Best, T.M.; Stimpfl, J.N.; He, F.; Zuo, L. Molecular characterization of redox mechanisms in allergic asthma. Ann. Allergy Asthma. Immunol. 2014, 113, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Page, L.K.; Staples, K.J.; Spalluto, C.M.; Watson, A.; Wilkinson, T. Influence of Hypoxia on the Epithelial-Pathogen Interactions in the Lung: Implications for Respiratory Disease. Front. Immunol. 2021, 12, 653969. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, A.; Broadfield, E.; Lewis, S.; Lawson, N.; Britton, J. Amino acids and asthma: A case-control study. Eur. Respir. J. 2004, 23, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.S.; Dahlin, A.; McGeachie, M.J.; Qiu, W.; Sordillo, J.; Wan, E.S.; Wu, A.C.; Lasky-Su, J. Asthma Metabolomics and the Potential for Integrative Omics in Research and the Clinic. Chest 2017, 151, 262–277. [Google Scholar] [CrossRef]
- Chang, W.K.; Yang, K.D.; Shaio, M.F. Lymphocyte proliferation modulated by glutamine: Involved in the endogenous redox reaction. Clin. Exp. Immunol. 1999, 117, 482–488. [Google Scholar] [CrossRef] [PubMed]
- van Hall, G.; van der Vusse, G.J.; Söderlund, K.; Wagenmakers, A.J. Deamination of amino acids as a source for ammonia production in human skeletal muscle during prolonged exercise. J. Physiol. 1995, 489 Pt 1, 251–261. [Google Scholar] [CrossRef]
- Busque, S.M.; Stange, G.; Wagner, C.A. Dysregulation of the glutamine transporter Slc38a3 (SNAT3) and ammoniagenic enzymes in obese, glucose-intolerant mice. Cell Physiol. Biochem 2014, 34, 575–589. [Google Scholar] [CrossRef]
- Sackesen, C.; Ercan, H.; Dizdar, E.; Soyer, O.; Gumus, P.; Tosun, B.N.; Büyüktuncer, Z.; Karabulut, E.; Besler, T.; Kalayci, O. A comprehensive evaluation of the enzymatic and nonenzymatic antioxidant systems in childhood asthma. J. Allergy Clin. Immunol. 2008, 122, 78–85. [Google Scholar] [CrossRef]
- Ko, H.M.; Kang, N.I.; Kim, Y.S.; Lee, Y.M.; Jin, Z.W.; Jung, Y.J.; Im, S.Y.; Kim, J.H.; Shin, Y.H.; Cho, B.H.; et al. Glutamine preferentially inhibits T-helper type 2 cell-mediated airway inflammation and late airway hyperresponsiveness through the inhibition of cytosolic phospholipase A(2) activity in a murine asthma model. Clin. Exp. Allergy 2008, 38, 357–364. [Google Scholar] [CrossRef]
- Bremer, J. Carnitine--metabolism and functions. Physiol. Rev. 1983, 63, 1420–1480. [Google Scholar] [CrossRef]
- Kertys, M.; Grendar, M.; Kosutova, P.; Mokra, D.; Mokry, J. Plasma based targeted metabolomic analysis reveals alterations of phosphatidylcholines and oxidative stress markers in guinea pig model of allergic asthma. Biochim. Biophys Acta Mol. Basis Dis. 2020, 1866, 165572. [Google Scholar] [CrossRef] [PubMed]
- Asilsoy, S.; Bekem, O.; Karaman, O.; Uzuner, N.; Kavukçu, S. Serum total and free carnitine levels in children with asthma. World J. Pediatr. 2009, 5, 60–62. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adduct | Name | VIP | Fold Change | p-Value | PPM |
|---|---|---|---|---|---|
| [M + H] + | Carnitine | 2.200291616 | 0.338584131 | 0.000426347 | 2.541680077 |
| [M + H] + | Glu-Gly-Arg | 1.818933829 | 4.591986615 | 0.000977944 | 0.869071719 |
| [M + H] + | Palmitoyl sphingomyelin | 4.642574354 | 0.476560728 | 0.001748888 | 0.153548353 |
| [M + Na] + | Lactose | 3.773556063 | 3.281279376 | 0.00259977 | 1.619460097 |
| [M + H] + | d-lactose | 1.857280581 | 2.851001893 | 0.003333963 | 3.055191786 |
| [M + H-H2O] + | Melibiose | 1.593316801 | 2.778378355 | 0.004764936 | 1.882063955 |
| [M + H-NH3] + | d-glutamine | 2.036482003 | 0.587719538 | 0.006265209 | 2.365700892 |
| [M + H] + | Glycerophosphocholine | 4.450704967 | 0.645349102 | 0.011104682 | 1.146858383 |
| [M + H] + | 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid | 9.213222731 | 1.284113383 | 0.015014811 | 0.536893891 |
| [M + H] + | Phosphorylcholine | 4.109895791 | 0.425042372 | 0.016706732 | 1.511398798 |
| [M + H] + | Pro-Trp | 1.571671351 | 0.757490368 | 0.017565433 | 1.702405805 |
| [M + H] + | 1-hexadecyl-2-(9z-octadecenoyl)-sn-glycero-3-phosphocholine | 3.02416122 | 0.570834294 | 0.032006053 | 1.19605325 |
| [M − H] − | (2e,6e,10e)-13-[(2r)-6-hydroxy-2,8-dimethyl-3,4-dihydrochromen-2-yl]-2,6,10-trimethyltrideca-2,6,10-trienoic acid | 1.548347947 | 6.564924847 | 4.61073 × 10−7 | 2.932669011 |
| [M − H] − | Mitragynine | 1.548364808 | 4.037763655 | 1.42082 × 10−6 | 2.99940101 |
| [M − H-HF] − | 5-heptenoic acid, 7-[(1r,2r,3s,5s)-2-[(1e,3s)-3-(2,3-dihydro-1h-inden-2-yl)-3-hydroxy-1-propen-1-yl]-3-fluoro-5-hydroxycyclopentyl]-, (5z)- | 1.753276464 | 6.597303608 | 1.60723 × 10−6 | 1.231379272 |
| [M − H] − | 2,2’-methylene-bis(6-tert-butyl)-4-ethylphenol | 7.255319656 | 687.7421105 | 1.47943 × 10−5 | 0.753989673 |
| [M + Hac-H] − | (2-{[3-hydroxy-2-tetradecanamidooctadec-4-en-1-yl phosphonato]oxy}ethyl)trimethylazanium | 1.744859227 | 0.101001266 | 8.2167 × 10−5 | 1.28624483 |
| [M − H] − | Mestranol | 1.55124963 | 3.498222121 | 0.000134048 | 4.974410509 |
| [M − H] − | Rauwolscine | 1.590991455 | 3.222450611 | 0.000858525 | 2.253295815 |
| [M − H] − | 1-hexadecanoyl-2-(9z-octadecenoyl)-sn-glycero-3-phospho-(1’-myo-inositol) | 2.674924291 | 0.46203907 | 0.000883256 | 1.278391498 |
| [M + CH3COOH-H] − | Sm d34:1 | 3.293558365 | 0.107293836 | 0.001682566 | 0.614405172 |
| [M − H] − | Pi 36:2 | 2.828081935 | 0.509520417 | 0.002043878 | 0.039202438 |
| [M − H] − | Cis,cis-muconic acid | 19.10365688 | 2.99791356 | 0.002282177 | 1.699372541 |
| [M − H] − | 2-oleoyl-1-stearoyl-sn-glycero-3-phosphoserine | 1.704491502 | 0.50233312 | 0.004191965 | 0.25523065 |
| [M − H] − | Pi(16:0e/15-hete) | 1.704070924 | 0.585164501 | 0.004289139 | 1.123702214 |
| [M − H] − | Myo-inositol | 1.930136424 | 0.763514117 | 0.00430323 | 0.431600743 |
| [M + Hac-H] − | Pc(16:1e/9-hode) | 2.134918492 | 1.124601576 | 0.004327026 | 1.399094575 |
| [M − H] − | 2-arachidonoyl-1-palmitoyl-sn-glycero-3-phosphoethanolamine | 1.960916534 | 0.619651975 | 0.004474611 | 3.180978267 |
| [M − H] − | 3-hydroxy-3-methylglutaric acid | 1.910771219 | 1.879446513 | 0.007537124 | 1.331120169 |
| [M − H] − | Glutamic acid | 2.692179409 | 0.642050185 | 0.008620487 | 2.04730699 |
| [M − H] − | N-acetyl-l-aspartic acid | 2.207757039 | 0.654700304 | 0.008774719 | 2.833624788 |
| [M − H] − | Pi 38:4 | 2.493421846 | 0.588702225 | 0.010326359 | 2.236373454 |
| [M − H] − | Pi 34:2 | 2.396638201 | 0.611586826 | 0.011323699 | 1.511310532 |
| [M − H] − | 3,4-dihydroxyhydrocinnamic acid | 2.448942044 | 2.368625638 | 0.012071806 | 3.876790668 |
| [M − H] − | Dl-lactate | 2.696831025 | 0.813506534 | 0.021585957 | 5.290769196 |
| [M − H] − | 1-stearoyl-2-linoleoyl-sn-glycero-3-phosphoethanolamine | 4.046902217 | 0.535272448 | 0.027432183 | 0.476942516 |
| [M − H] − | Pe 32:1 | 2.224649115 | 0.60913988 | 0.031056552 | 0.638952989 |
| [M − H] − | (2-aminoethoxy)[3-[hexadec-1-en-1-yloxy]-2-[icosa-5.8.11.14-tetraenoyloxy]propoxy]phosphinic acid | 5.285454225 | 0.616442722 | 0.031251857 | 0.949210223 |
| [M − H] − | 1-palmitoyl-2-oleoyl-phosphatidylglycerol | 5.322414542 | 0.576304139 | 0.031944236 | 2.292568905 |
| [M − H] − | Pe(18:1e/12-hete) | 2.427631126 | 0.623348149 | 0.034032449 | 5.956456416 |
| [M − H] − | 5’-phosphoribosyl-5-amino-4-imidazolecarboxamide (aicar) | 1.694809852 | 1.197983127 | 0.036763439 | 4.262055988 |
| [M − H] − | 2-linoleoyl-1-palmitoyl-sn-glycero-3-phosphoethanolamine | 3.019191108 | 0.654770708 | 0.036864178 | 1.513458381 |
| [M − H] − | Pe(16:1e/15-hete) | 2.007563238 | 0.592051877 | 0.042176928 | 2.41362126 |
| [M − H] − | (2-aminoethoxy)[2-[docosa-4.7.10.13.16.19-hexaenoyloxy]-3-[hexadec-1-en-1-yloxy]propoxy]phosphinic acid | 4.5379477 | 0.661536657 | 0.044480761 | 0.169100171 |
| [M − H] − | d-mannose | 2.054375169 | 2.637354787 | 0.048367452 | 2.341916416 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, F.; Yu, J.; Lai, T.; Luo, L.; Zhang, W. The Combination of Bioinformatics Analysis and Untargeted Metabolomics Reveals Potential Biomarkers and Key Metabolic Pathways in Asthma. Metabolites 2023, 13, 25. https://doi.org/10.3390/metabo13010025
Huang F, Yu J, Lai T, Luo L, Zhang W. The Combination of Bioinformatics Analysis and Untargeted Metabolomics Reveals Potential Biomarkers and Key Metabolic Pathways in Asthma. Metabolites. 2023; 13(1):25. https://doi.org/10.3390/metabo13010025
Chicago/Turabian StyleHuang, Fangfang, Jinjin Yu, Tianwen Lai, Lianxiang Luo, and Weizhen Zhang. 2023. "The Combination of Bioinformatics Analysis and Untargeted Metabolomics Reveals Potential Biomarkers and Key Metabolic Pathways in Asthma" Metabolites 13, no. 1: 25. https://doi.org/10.3390/metabo13010025
APA StyleHuang, F., Yu, J., Lai, T., Luo, L., & Zhang, W. (2023). The Combination of Bioinformatics Analysis and Untargeted Metabolomics Reveals Potential Biomarkers and Key Metabolic Pathways in Asthma. Metabolites, 13(1), 25. https://doi.org/10.3390/metabo13010025