

Identification of Plant-Derived Bioactive Compounds Using Affinity Mass Spectrometry and Molecular Networking

 ,
,
Abstract
:1. Introduction
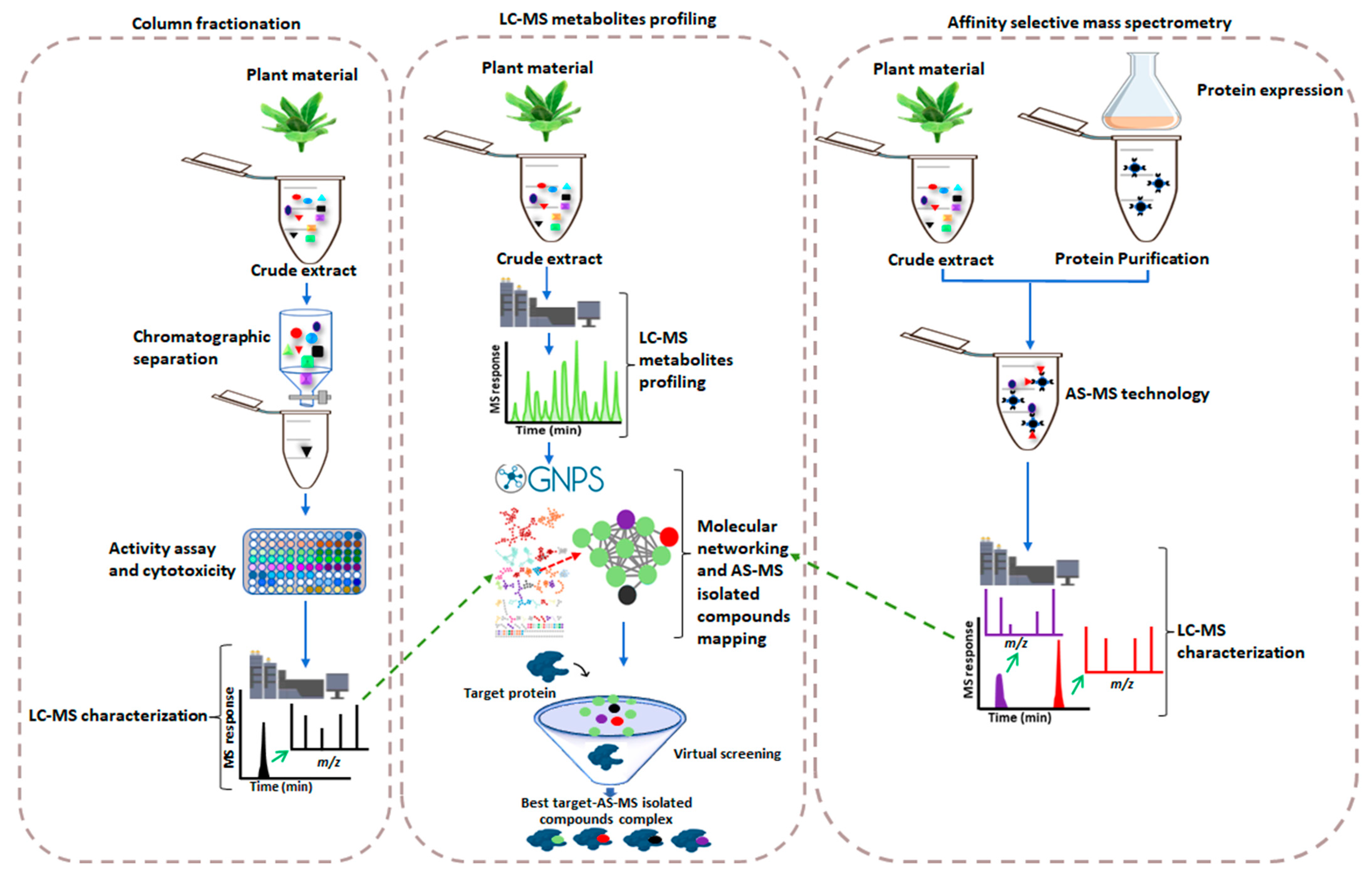
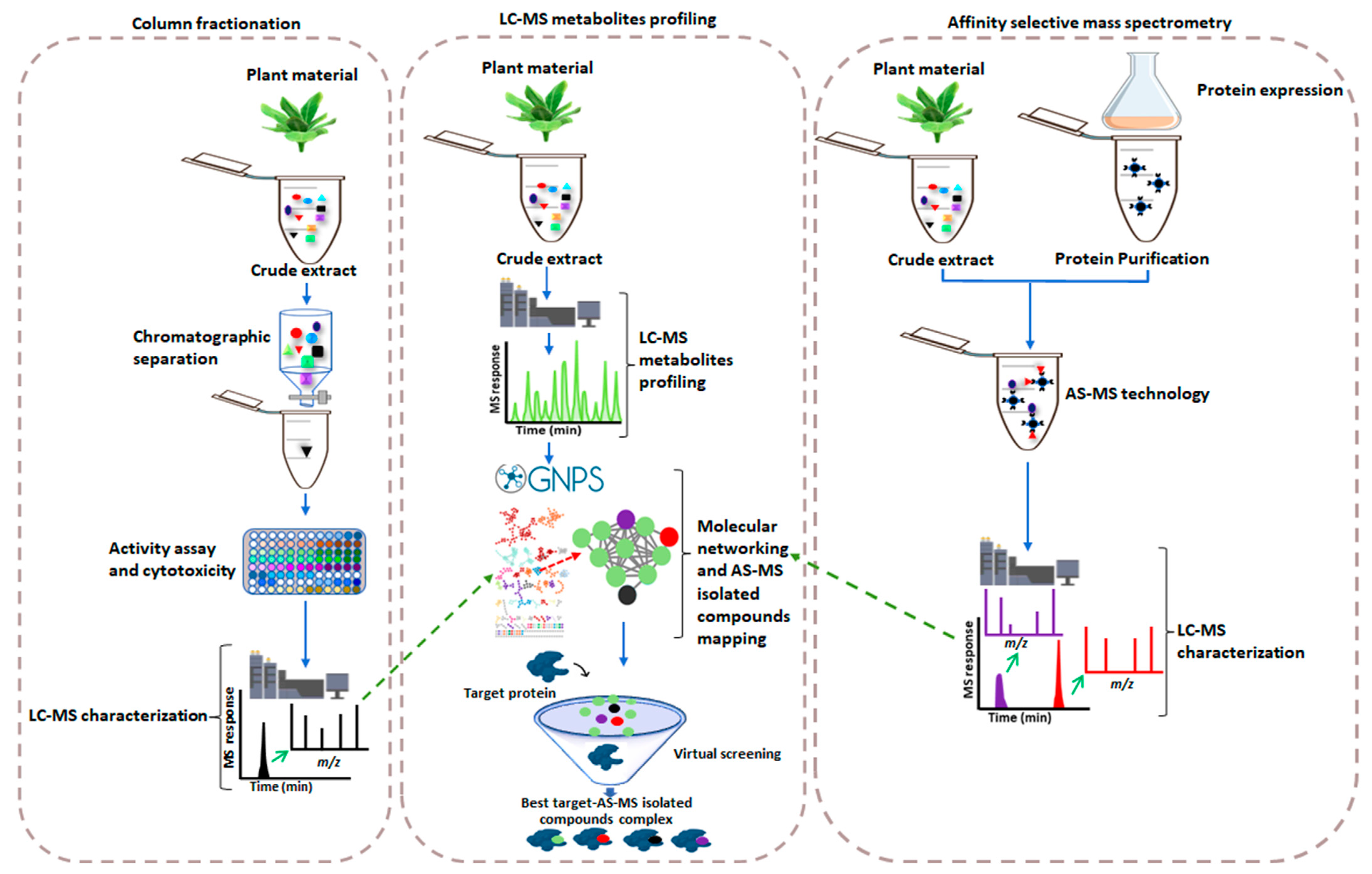
2. Affinity Selection-Mass Spectrometry-Based Drug Discovery versus the Conventional Method
Metabolite Profiling Meeting AS-MS
3. Concluding Remarks
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Lahlou, M. The Success of Natural Products in Drug Discovery. J. Pharm. Pharmacol. 2013, 4, 17–31. [Google Scholar] [CrossRef]
- Muchiri, R.N.; van Breemen, R.B. Affinity selection–mass spectrometry for the discovery of pharmacologically active compounds from combinatorial libraries and natural products. J. Mass. Spectrom. 2021, 56, e4647. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.W.; Balunas, M.J.; Chai, H.B.; Kinghorn, A.D. Drug discovery from natural sources. AAPS J. 2006, 8, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–221. [Google Scholar] [CrossRef] [PubMed]
- Rishton, G.M. Natural products as a robust source of new drugs and drug leads past successes and present-day issues. Am. J. Cardiol. 2008, 101, 43D–49D. [Google Scholar] [CrossRef]
- Dias, D.A.; Urban, S.; Roessner, U. A historical overview of natural products in drug discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef]
- Lu, Y.; Qin, S.; Zhang, B.; Dai, A.; Cai, X.; Ma, M.; Gao, Z.; Yang, D.; Stevens, R.C.; Jacobson, K.A.; et al. Accelerating the throughput of affinity mass spectrometry-based ligand screening towards a G protein-coupled receptor. Anal. Chem. 2019, 91, 8162–8169. [Google Scholar] [CrossRef]
- Rao, S.V.; Srinivas, K. Modern drug discovery process: An in silico approach. JBSA 2011, 2, 89–94. [Google Scholar]
- Louie, K.B. Comprehensive Natural Products III: Mass Spectrometry for Natural Product Discovery; Lawrence Berkeley National Laboratory (LBNL): Berkeley, CA, USA, 2020; pp. 63–306. [Google Scholar]
- Annis, A.; Chuang, C.C.; Nazef, N. Methods and Principles in Medicinal Chemistry Mass Spectrometry in Medicinal Chemistry || ALIS: An Affinity Selection–Mass Spectrometry System for the Discovery and Characterization of Protein–Ligand Interactions; Wiley-VCH Verlag GmbH & Co.KgaA: Hoboken, NJ, USA, 2007; Chapter 3,; pp. 121–156. Available online: https://onlinelibrary.wiley.com/doi/book/10.1002/9783527610907 (accessed on 8 September 2022).
- Annis, D.; Athanasopoulos, J.; Curran, P.; Felsch, J.; Kalghatgi, K.; Lee, W.; Nash, H.; Orminati, J.-P.; Rosner, K.; Shipps, G.; et al. An affinity selection-mass spectrometry method for the identification of small molecule ligands from self-encoded combinatorial libraries—Discovery of a novel antagonist of E-coli dihydrofolate reductase. Int. J. Mass Spectrom. 2004, 238, 77–83. [Google Scholar]
- Zehender, H.; Mayr, L.M. Application of high-throughput affinity-selection mass spectrometry for screening of chemical compound libraries in lead discovery. Expert Opin. Drug Discov. 2007, 2, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Muchiri, R.N.; van Breemen, R.B. Drug discovery from natural products using affinity selection-mass spectrometry. Drug Discov. Today Technol. 2021, 40, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Mak, T.; Rossojohn, J.; Littler, D.E.; Liu, M.; Quinn, R.J. Collision-induced affinity selection mass spectrometry for identification of ligands. ACS Bio. Med. Chem. Au. 2022. [Google Scholar] [CrossRef]
- Van Breemen, R.B.; Muchiri, R.N.; Bates, T.A.; Weinstein, J.B.; Leier, H.C.; Farley, S.; Tafesse, F.G. Cannabinoids Block Cellular Entry of SARS-CoV-2 and the Emerging Variants. J. Nat. Prod. 2022, 85, 176–184. [Google Scholar] [CrossRef]
- Tamara, S.; den Boer, M.A.; Heck, A.J.R. High-resolution native mass spectrometry. Chem Rev. 2022, 122, 7269–7326. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, C.; Annis, D. Affinity selection-mass spectrometry and its emerging application to the high throughput screening of G protein-coupled receptors. Comb. Chem. High Throughput Screen. 2008, 11, 427–438. [Google Scholar] [CrossRef]
- Prudent, R.; Annis, D.A.; Dandliker, P.J.; Ortholand, J.Y.; Roche, D. Exploring new targets and chemical space with affinity selection-mass spectrometry. Nat. Rev. Chem. 2020, 5, 62–71. [Google Scholar] [CrossRef]
- Motoyaji, T. Revolution of small molecule drug discovery by affinity selection-mass spectrometry technology. Chem. Pharm. Bull. 2020, 68, 191–193. [Google Scholar] [CrossRef]
- Zhu, D.; Su, H.; Ke, C.; Tang, C.; Witt, M.; Quinn, R.J.; Xu, Y.; Liu, J.; Ye, Y. Efficient discovery of potential inhibitors for SARS-CoV-2 3C-like protease from herbal extracts using a native MS-based affinity-selection method. J. Pharm. Biomed. Anal. 2022, 209, 114538. [Google Scholar] [CrossRef]
- Fu, Y.; Mo, H.-Y.; Gao, W.; Hong, J.-Y.; Lu, J.; Li, P.; Chen, J. Affinity selection-based two-dimensional chromatography coupled with high-performance liquid chromatography-mass spectrometry for discovering xanthine oxidase inhibitors from Radix Salviae miltiorrhizae. Anal. Bioanal. Chem. 2014, 406, 4987–4995. [Google Scholar] [CrossRef]
- Fu, X.; Wang, Z.; Li, L.; Dong, S.; Li, Z.; Jiang, Z.; Shui, W. Novel chemical ligands to ebola virus and marburg virus nucleoproteins identified by combining affinity mass spectrometry and metabolomics approaches. Sci. Rep. 2016, 6, 29680. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kim, U.; Liu, J.; Cheng, C.; Wu, W.; Guo, S.; Bai, G. Comprehensive TCM molecular networking based on MS/MS in silico spectra with the integration of virtual screening and affinity MS screening for discovering functional ligands from natural herbs. Anal. Bioanal. Chem. 2019, 411, 5785–5797. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liang, H.; Cao, H.; Zhang, B.; Li, J.; Wang, W.; Qin, S.; Wang, F.; Xuan, L.; Lai, L.; et al. Efficient ligand discovery from natural herbs by integrating virtual screening, affinity mass spectrometry and targeted metabolomics. Analyst 2019, 144, 2881–2890. [Google Scholar] [CrossRef]
- Tao, Y.; Chen, Z.; Zhang, Y.; Wang, Y.; Cheng, Y. Immobilised magnetic beads based multi-target affinity selection coupled with high performance liquid chromatography–mass spectrometry for screening anti-diabetic compounds from a Chinese medicine “Tang-Zhi-Qing”. J. Pharm. Biomed. Anal. 2013, 78–79, 190–201. [Google Scholar] [CrossRef]
- Imaduwage, K. High-Throughput Screening (HTS) of Potential Lead Compounds for Target Proteins with no False Identifications Using LC/MS. Bachelor’s Thesis, University of Kansas, Lawrence, KS, USA, 2017. [Google Scholar] [CrossRef]
- Henrich, C.J.; Beutler, J.A. Matching the power of high throughput screening to the chemical diversity of natural products. Nat. Prod. Rep. 2013, 30, 1284. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Luzzatto-Knaan, T. Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, R.R.; Wang, M.; Nothias, L.-F.; van der Hooft, J.J.J.; Caraballo-Rodríguez, A.M.; Fox, E.; Dorrestein, P.C. Propagating annotations of molecular networks using in silico fragmentation. PLOS Comput. Biol. 2018, 14, e1006089. [Google Scholar] [CrossRef]
- Olivon, F.; Allard, P.M.; Koval, A.; Righi, D.; Genta-Jouve, G.; Neyts, J.; Apel, C.; Pannecouque, C.; Nothias, L.F.; Cachet, X.; et al. Bioactive natural products prioritisation using massive multi-informational molecular networks. ACS Chem. Biol. 2017, 12, 2644–2651. [Google Scholar] [CrossRef]
- Vyas, V. Virtual screening: A fast tool for drug design. Sci. Pharm. 2008, 76, 333–360. [Google Scholar] [CrossRef] [Green Version]
- Ingelheim, B. Virtual screening - What does it give us? Curr. Opin. Drug Discov. Dev. 2009, 12, 397–407. [Google Scholar]

{kind=link}
| Description | References | |
|---|---|---|
| 1. Affinity selection | ||
| Magnetic microbead affinity selection screening (MagMASS) | MagMASS is a solid-phase alternative that complements the solution-phase screening approaches. MagMASS involves tethering the target to magnetic microbeads, incubating the immobilized protein with a natural product mixture, using magnetism to separate the ligand-protein/bead complexes from unbound compounds, and then releasing the bound ligands for UHPLC-MS analysis. | [16] |
| Pulsed ultrafiltration (PUF) AS-MS. | PUF AS-MS screening begins with the incubation of a mixture of compounds, such as a natural product extract with a solution-phase macromolecular receptor (protein, enzyme, or RNA). After equilibrium is achieved, ultrafiltration is used to separate the large ligand-receptor complexes from the unbound low-mass compounds. Because large pore sizes enable faster ultrafiltration separation, the pore size of the ultrafiltration membrane should be as large as possible while still retaining the macromolecular receptor. | [16] |
| Collision-induced affinity selection mass spectrometry (CIAS-MS) | Collision-induced affinity selection mass spectrometry (CIAS-MS) is a new method that relies on the affinity between a protein and its ligand for the identification of ligands. | [15] |
| Size exclusion chromatography (SEC) AS-MS | SEC AS-MS is a solution-phase screening approach like PUF AS-MS that begins with the incubation of a mixture of possible ligands with a macromolecular receptor. After equilibrium is achieved, SEC is used to separate the large ligand-receptor complexes from smaller, unbound compounds. The high mass complexes elute first during SEC and are then denatured using an organic solvent to release the ligands for reversed-phase LC-MS analysis. | [16] |
| 2. Native MS | ||
| Bioassay-guided fractionation-MS | This involves the analysis and characterization of molecules whereby the native structural features of the analytes are retained as much as possible. It provides binding informationabout each compound towards the protein of interest. | [17] |
| Plant | Compound | Target | Docking | Main Results | References |
|---|---|---|---|---|---|
| Cannabis sativa |
|
| Cannabigerolic acid binds to the anallosteric site of S1 with −6.6 kcal/mol binding energy.Cannabidiolic acid also binds at the orthosteric site with −6.3 kcal/mol.THCA-A bind at the orthosteric site with −6.5 kcal/mol binding energy. | Bound to the spike protein thus preventing entry into the cell. | [16] |
| Radix salvia miltiorrhiza |
|
| No | Salvianolic acid C exhibited potent XOD inhibitory activity with an IC50 of 9.07 μM. | [22] |
| Scutellaria baicalensis |
|
| No | Three flavonoids were identified as potential noncovalent inhibitors against 3CLpro with IC50 values of 0.94, 3.02, and 0.84 µM, respectively. | [21] |
| Gancao (licorice root) |
|
| In silico docking analysis was employed to create a potential model for binding of GC7 and GC13 to EBOV nucleoprotein. | By combining affinity mass spectrometry and metabolomics approaches, two compounds were identified from a traditional Chinese medicine Gancao (licorice root) that binds to nucleoproteins (NPs). These two ligands, 18β-glycyrrhetinic acid, and licochalcone A were verified by defined compound mixture screens and further characterized with individual ligand binding assays. | [23] |
| Rhizoma atractylodis macrocephalaeRhizoma pinelliaeBulbus fritillariaRhizoma paridisRhizoma curcumaeFructus trichosanthisRhizoma dioscoreae bulbiferaeRadix sophorae flavescentisRadix ginsengRadix notoginsengRadix asparagi |
|
| For the docking analysis, ligands kurarinol, kurarinone, 20(s)-Rg3, and 20(s)-Rh2 were inserted into the GTP-binding pocket and the results demonstrated that kurarinol and kurarinone competed with GTP. | Molecular networking and virtual screening coupled with affinity selection-mass spectrometry discovered two compounds, kurarinol and kurarinone, were confirmed to interact with GTPase of Ras and were successfully identified from 11 traditional Chinese medicine (TCM) herbs. | [24] |
| Piper kadsuraPiper nigrumOphiopogon japonicusSalvia miltiorrhiza |
|
| Molecular docking studies were performed to create a docking model of HJ-4 interacting with the hydrophobic pocket in the C-lobe of the nucleoprotein. | Through affinity selection-mass spectrometry approach, three compounds isolated from Piper nigrum (HJ-1, HJ-4, and HJ-6) strongly promoted the formation of large nucleoprotein oligomers and reduced the protein thermal stability, and docking studies were performed to show the interaction of HJ-4 to EBOV nucleoprotein. | [25] |
| Glycyrrhiza inflata |
|
| No | Small molecule ligands to the spike protein were discovered in extracts of the licorice species, Glycyrrhiza inflata. In particular, two hits were detected during screening of Glycyrrhiza inflata, and hit one was identified as licochalcone A while hit 2 corresponded to licoflavone B and glyinflanin K. However, in the absence of authentic standards, the conformation of this ligand (hit 2) is still ongoing. | [3] |
| Rabdosia rubescens | Oridonin | Nsp9 Protein | No | A known SARS-CoV-2 Nsp9 ligand, oridonin, was successfully detected when it was mixed with Nsp9 | [15] |
| Tang-zhi-qing |
|
| No | Through the use of multiple target-immobilized magnetic beads coupled with high-performance liquid chromatography–mass spectrometry, five active compounds, namely, 2,3,4,6-tetra-O-galloyl-d-glucose, 1,2,3,4-tetra-O-galloyl-d-glucose, 1,2,3,4,6-penta-O-galloyl-d-glucose, quercetin-3-O-β-d-glucuronide, and quercetin-3-O-β-d-glucoside, were identified and their activities were validated by conventional inhibitory assay. | [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramatapa, T.; Msobo, A.; Maphari, P.W.; Ncube, E.N.; Nogemane, N.; Mhlongo, M.I. Identification of Plant-Derived Bioactive Compounds Using Affinity Mass Spectrometry and Molecular Networking. Metabolites 2022, 12, 863. https://doi.org/10.3390/metabo12090863
Ramatapa T, Msobo A, Maphari PW, Ncube EN, Nogemane N, Mhlongo MI. Identification of Plant-Derived Bioactive Compounds Using Affinity Mass Spectrometry and Molecular Networking. Metabolites. 2022; 12(9):863. https://doi.org/10.3390/metabo12090863
Chicago/Turabian StyleRamatapa, Thabo, Anathi Msobo, Pfano W. Maphari, Efficient N. Ncube, Noluyolo Nogemane, and Msizi I. Mhlongo. 2022. "Identification of Plant-Derived Bioactive Compounds Using Affinity Mass Spectrometry and Molecular Networking" Metabolites 12, no. 9: 863. https://doi.org/10.3390/metabo12090863
APA StyleRamatapa, T., Msobo, A., Maphari, P. W., Ncube, E. N., Nogemane, N., & Mhlongo, M. I. (2022). Identification of Plant-Derived Bioactive Compounds Using Affinity Mass Spectrometry and Molecular Networking. Metabolites, 12(9), 863. https://doi.org/10.3390/metabo12090863