
Effect of Expression of Nuclear-Encoded Cytochrome C Oxidase Subunit 4 Isoforms on Metabolic Profiles of Glioma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Glioma Cell Lines
2.2. Sample Preparation
2.3. UPLC-MS/MS
2.4. Bioinformatics
2.5. Data Extraction and Compound Identification
2.6. Metabolite Quantification and Data Normalization
2.7. Statistical Analysis
3. Results
3.1. Effects of COX4 Isoforms on the Glioma Cell Metabolome
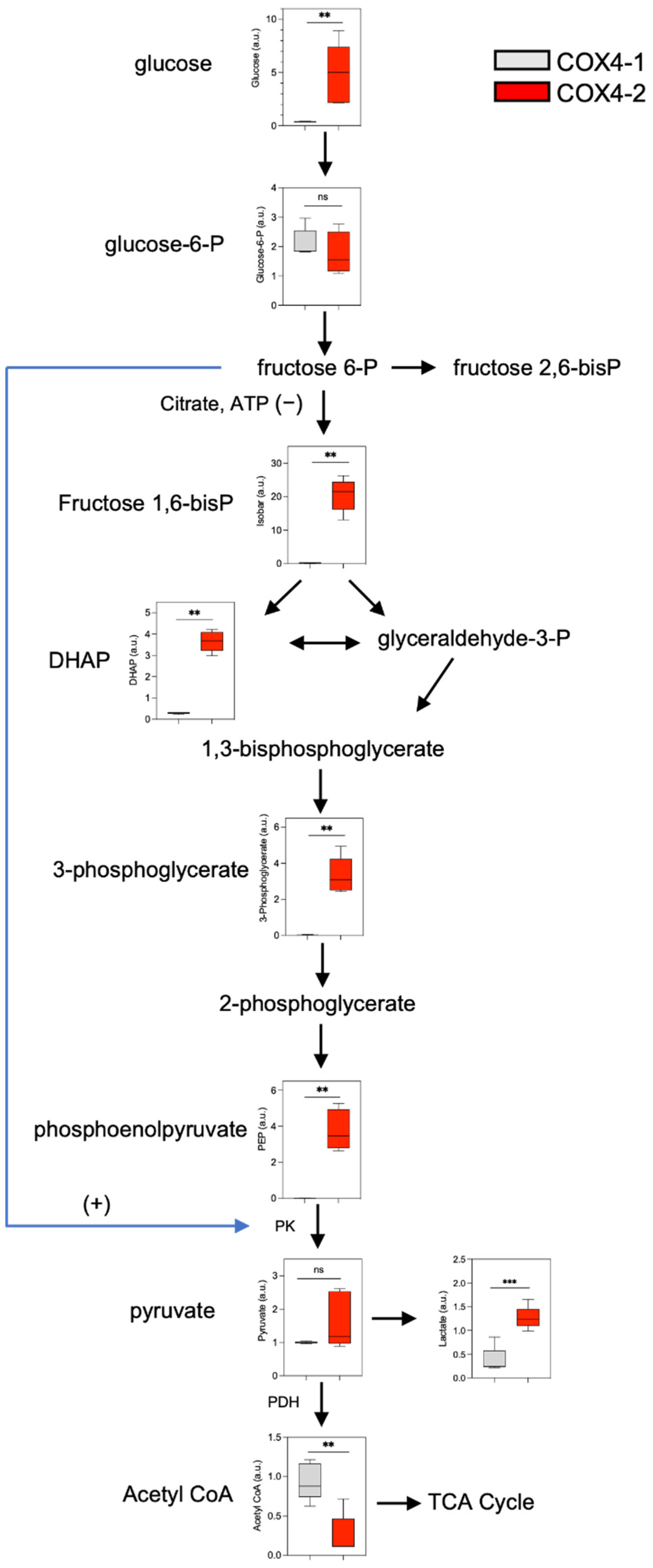
3.2. Analysis of Glucose Metabolism in Glioma Cells Overexpressing COX4 Isoforms
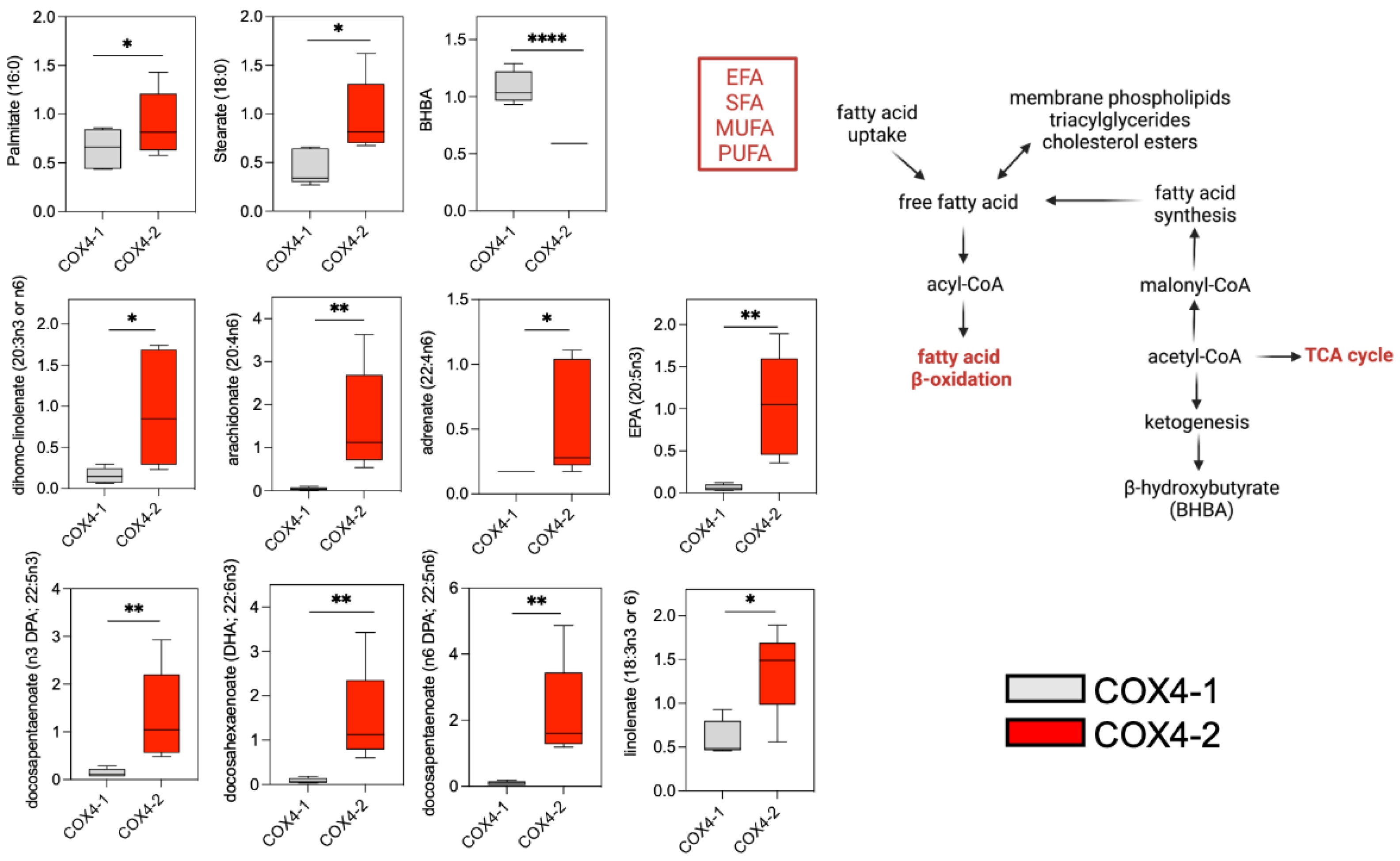
3.3. Analysis of Fatty Acid Metabolism in Glioma Cells Overexpressing COX4 Isoforms
3.4. Analysis of Glutathione Metabolism in Glioma Cells Overexpressing COX4 Isoforms
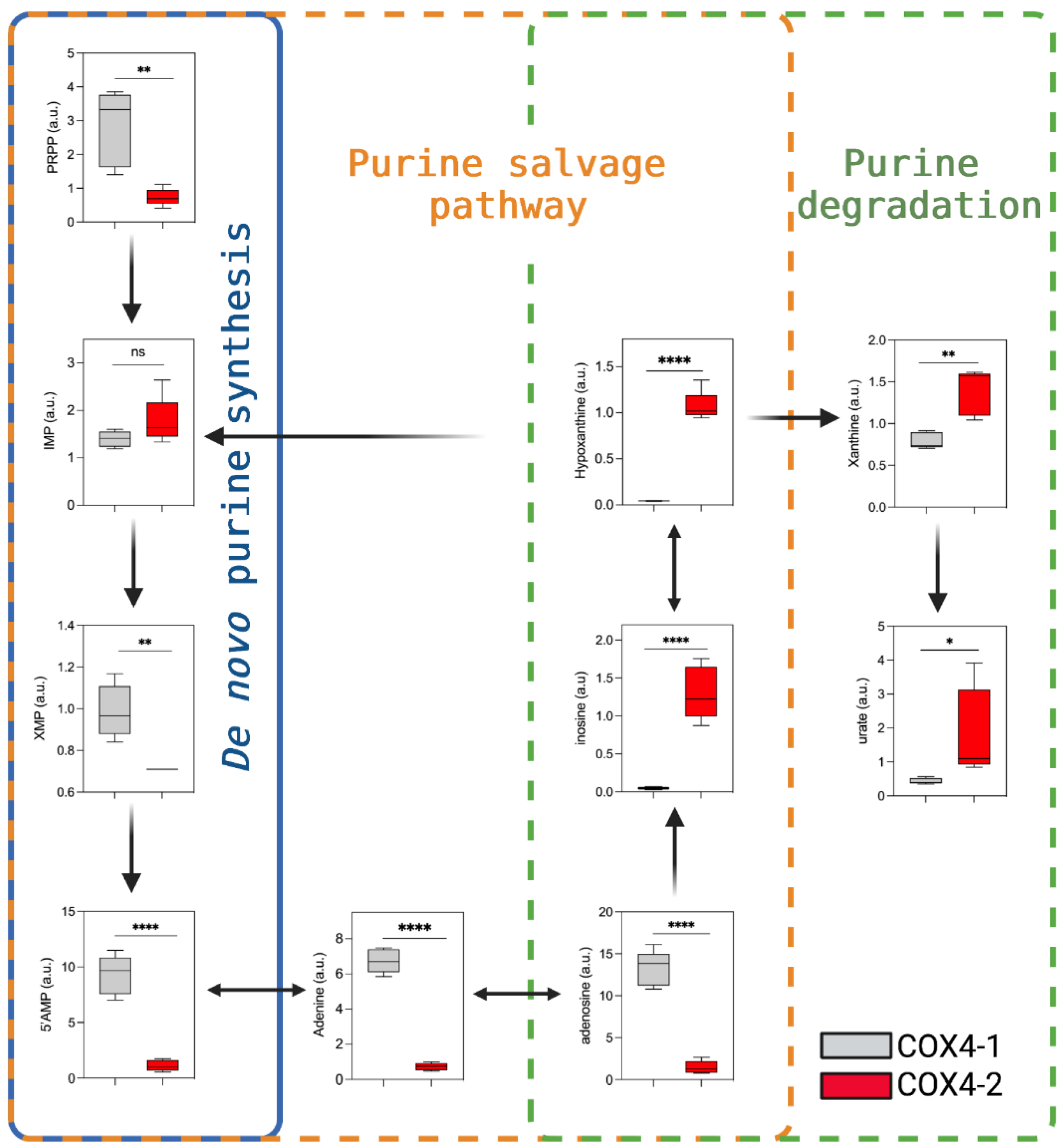
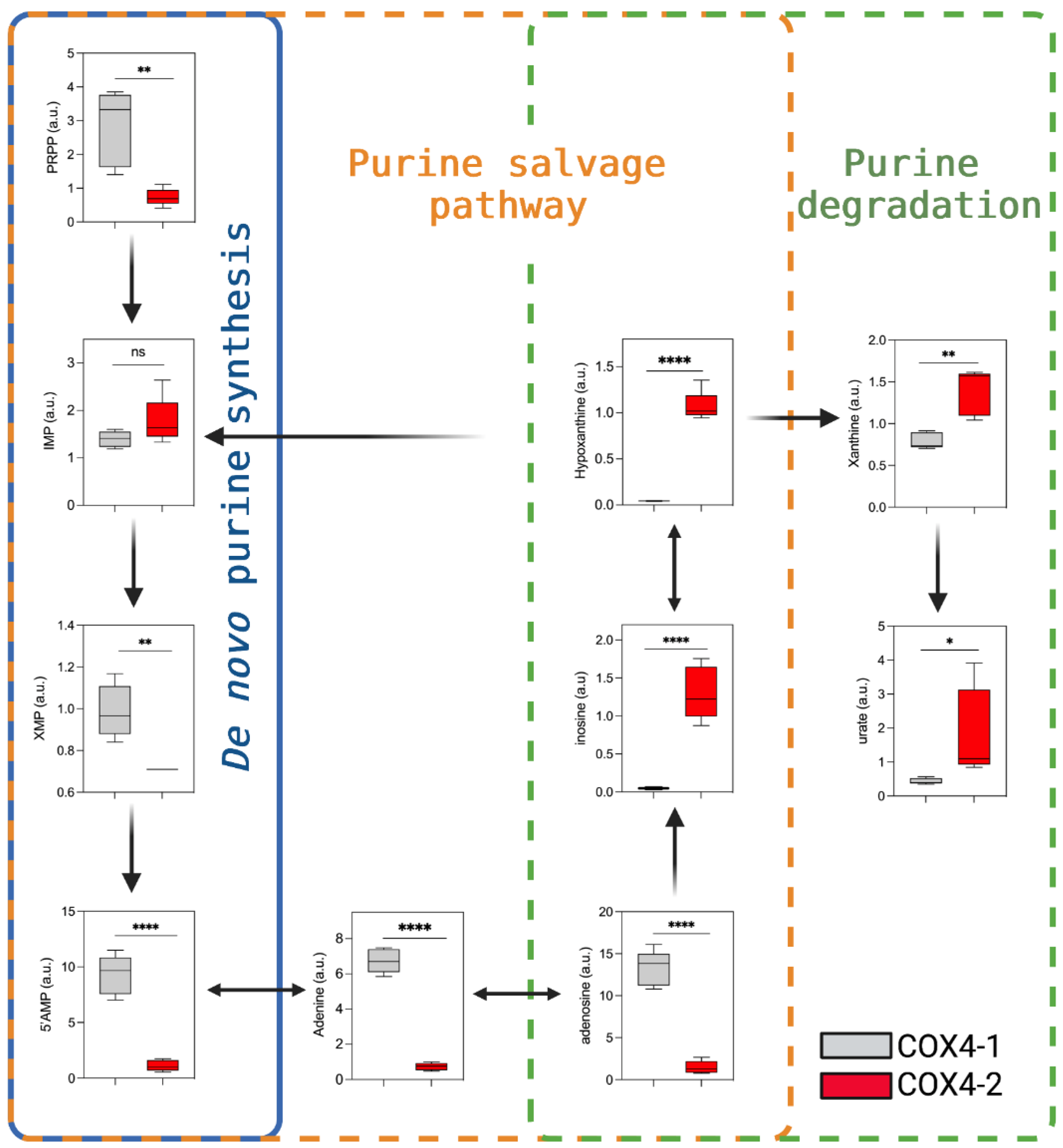
3.5. Analysis of Purine Nucleotide Synthesis in Glioma Cells Overexpressing COX4 Isoforms
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oliva, C.R.; Moellering, D.R.; Gillespie, G.Y.; Griguer, C.E. Acquisition of chemoresistance in gliomas is associated with increased mitochondrial coupling and decreased ROS production. PLoS ONE 2011, 6, e24665. [Google Scholar] [CrossRef] [PubMed]
- Oliva, C.R.; Nozell, S.E.; Diers, A.; McClugage, S.G., 3rd; Sarkaria, J.N.; Markert, J.M.; Darley-Usmar, V.M.; Bailey, S.M.; Gillespie, G.Y.; Landar, A.; et al. Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain. J. Biol. Chem. 2010, 285, 39759–39767. [Google Scholar] [CrossRef] [PubMed]
- Oliva, C.R.; Markert, T.; Gillespie, G.Y.; Griguer, C.E. Nuclear-encoded cytochrome c oxidase subunit 4 regulates BMI1 expression and determines proliferative capacity of high-grade gliomas. Oncotarget 2015, 6, 4330–4344. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Oliva, C.R.; Flor, S.; Goswami, P.C.; Griguer, C.E. Cytochrome c oxidase mediates labile iron level and radioresistance in glioblastoma. Free Radic. Biol. Med. 2022, 185, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Oliva, C.R.; Ali, M.Y.; Flor, S.; Griguer, C.E. COX4-1 promotes mitochondrial supercomplex assembly and limits reactive oxide species production in radioresistant GBM. Cell Stress 2022, 6, 45–60. [Google Scholar] [CrossRef]
- Evans, A.M.; DeHaven, C.D.; Barrett, T.; Mitchell, M.; Milgram, E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal. Chem. 2009, 81, 6656–6667. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Maus, A.; Peters, G.J. Glutamate and alpha-ketoglutarate: Key players in glioma metabolism. Amino Acids 2017, 49, 21–32. [Google Scholar] [CrossRef]
- Killian, J.K.; Kim, S.Y.; Miettinen, M.; Smith, C.; Merino, M.; Tsokos, M.; Quezado, M.; Smith, W.I., Jr.; Jahromi, M.S.; Xekouki, P.; et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013, 3, 648–657. [Google Scholar] [CrossRef]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef]
- Rodriguez-Enriquez, S.; Hernandez-Esquivel, L.; Marin-Hernandez, A.; El Hafidi, M.; Gallardo-Perez, J.C.; Hernandez-Resendiz, I.; Rodriguez-Zavala, J.S.; Pacheco-Velazquez, S.C.; Moreno-Sanchez, R. Mitochondrial free fatty acid beta-oxidation supports oxidative phosphorylation and proliferation in cancer cells. Int. J. Biochem. Cell Biol. 2015, 65, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Dong, X.; Xiao, L.; Tan, Z.; Luo, X.; Yang, L.; Li, W.; Shi, F.; Li, Y.; Zhao, L.; et al. CPT1A-mediated fatty acid oxidation promotes cell proliferation via nucleoside metabolism in nasopharyngeal carcinoma. Cell Death Dis. 2022, 13, 331. [Google Scholar] [CrossRef] [PubMed]
- Mierziak, J.; Burgberger, M.; Wojtasik, W. 3-Hydroxybutyrate as a Metabolite and a Signal Molecule Regulating Processes of Living Organisms. Biomolecules 2021, 11, 402. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Du, S.; Du, Y.; Ren, J.; Ying, G.; Yan, Z. Glutathione reductase mediates drug resistance in glioblastoma cells by regulating redox homeostasis. J. Neurochem. 2018, 144, 93–104. [Google Scholar] [CrossRef]
- Desideri, E.; Ciccarone, F.; Ciriolo, M.R. Targeting Glutathione Metabolism: Partner in Crime in Anticancer Therapy. Nutrients 2019, 11, 1926. [Google Scholar] [CrossRef]
- Olivier, C.; Oliver, L.; Lalier, L.; Vallette, F.M. Drug Resistance in Glioblastoma: The Two Faces of Oxidative Stress. Front. Mol. Biosci. 2020, 7, 620677. [Google Scholar] [CrossRef]
- Liu, Y.; Hyde, A.S.; Simpson, M.A.; Barycki, J.J. Emerging regulatory paradigms in glutathione metabolism. Adv. Cancer Res. 2014, 122, 69–101. [Google Scholar] [CrossRef]
- Soga, T.; Baran, R.; Suematsu, M.; Ueno, Y.; Ikeda, S.; Sakurakawa, T.; Kakazu, Y.; Ishikawa, T.; Robert, M.; Nishioka, T.; et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 2006, 281, 16768–16776. [Google Scholar] [CrossRef]
- Hu, J.; Locasale, J.W.; Bielas, J.H.; O’Sullivan, J.; Sheahan, K.; Cantley, L.C.; Vander Heiden, M.G.; Vitkup, D. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat. Biotechnol. 2013, 31, 522–529. [Google Scholar] [CrossRef]
- Zhou, W.; Yao, Y.; Scott, A.J.; Wilder-Romans, K.; Dresser, J.J.; Werner, C.K.; Sun, H.; Pratt, D.; Sajjakulnukit, P.; Zhao, S.G.; et al. Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat. Commun. 2020, 11, 3811. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B.; Huttemann, M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 2015, 24, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B. Regulation of cytochrome c oxidase contributes to health and optimal life. World J. Biol. Chem. 2020, 11, 52–61. [Google Scholar] [CrossRef]
- Poyton, R.O.; McEwen, J.E. Crosstalk between nuclear and mitochondrial genomes. Annu. Rev. Biochem. 1996, 65, 563–607. [Google Scholar] [CrossRef] [PubMed]
- Cunatova, K.; Reguera, D.P.; Vrbacky, M.; Fernandez-Vizarra, E.; Ding, S.; Fearnley, I.M.; Zeviani, M.; Houstek, J.; Mracek, T.; Pecina, P. Loss of COX4I1 Leads to Combined Respiratory Chain Deficiency and Impaired Mitochondrial Protein Synthesis. Cells 2021, 10, 369. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Warburg, O.; Gawehn, K.; Geissler, A.W. The transformation of embryonal metabolism in cancer metabolism. Z. Nat. B 1960, 15, 378–379. Available online: https://pubmed.ncbi.nlm.nih.gov/13855297 (accessed on 14 June 2022).
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Perspectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef]
- Anderson, N.M.; Li, D.; Peng, H.L.; Laroche, F.J.; Mansour, M.R.; Gjini, E.; Aioub, M.; Helman, D.J.; Roderick, J.E.; Cheng, T.; et al. The TCA cycle transferase DLST is important for MYC-mediated leukemogenesis. Leukemia 2016, 30, 1365–1374. [Google Scholar] [CrossRef]
- Bunik, V.I.; Mkrtchyan, G.; Grabarska, A.; Oppermann, H.; Daloso, D.; Araujo, W.L.; Juszczak, M.; Rzeski, W.; Bettendorff, L.; Fernie, A.R.; et al. Inhibition of mitochondrial 2-oxoglutarate dehydrogenase impairs viability of cancer cells in a cell-specific metabolism-dependent manner. Oncotarget 2016, 7, 26400–26421. [Google Scholar] [CrossRef] [PubMed]
- Abla, H.; Sollazzo, M.; Gasparre, G.; Iommarini, L.; Porcelli, A.M. The multifaceted contribution of alpha-ketoglutarate to tumor progression: An opportunity to exploit? Semin. Cell Dev. Biol. 2020, 98, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Atlante, S.; Visintin, A.; Marini, E.; Savoia, M.; Dianzani, C.; Giorgis, M.; Surun, D.; Maione, F.; Schnutgen, F.; Farsetti, A.; et al. Alpha-ketoglutarate dehydrogenase inhibition counteracts breast cancer-associated lung metastasis. Cell Death Dis. 2018, 9, 756. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, K.J.; Wilson, E.B.; Short, S.; Melcher, A.A.; Biggs, M.; Diakos, C.I.; Howell, V.M. Glycolysis and Fatty Acid Oxidation Inhibition Improves Survival in Glioblastoma. Front. Oncol. 2021, 11, 633210. [Google Scholar] [CrossRef]
- Prabhu, A.H.; Kant, S.; Kesarwani, P.; Ahmed, K.; Forsyth, P.; Nakano, I.; Chinnaiyan, P. Integrative cross-platform analyses identify enhanced heterotrophy as a metabolic hallmark in glioblastoma. Neuro. Oncol. 2019, 21, 337–347. [Google Scholar] [CrossRef]
- Grabacka, M.M.; Wilk, A.; Antonczyk, A.; Banks, P.; Walczyk-Tytko, E.; Dean, M.; Pierzchalska, M.; Reiss, K. Fenofibrate Induces Ketone Body Production in Melanoma and Glioblastoma Cells. Front. Endocrinol. 2016, 7, 5. [Google Scholar] [CrossRef]
- Maurer, G.D.; Brucker, D.P.; Bahr, O.; Harter, P.N.; Hattingen, E.; Walenta, S.; Mueller-Klieser, W.; Steinbach, J.P.; Rieger, J. Differential utilization of ketone bodies by neurons and glioma cell lines: A rationale for ketogenic diet as experimental glioma therapy. BMC Cancer 2011, 11, 315. [Google Scholar] [CrossRef]
- Patel, M.S.; Russell, J.J.; Gershman, H. Ketone-body metabolism in glioma and neuroblastoma cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7214–7218. [Google Scholar] [CrossRef]
- Poff, A.M.; Ari, C.; Arnold, P.; Seyfried, T.N.; D’Agostino, D.P. Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer. Int. J. Cancer. 2014, 135, 1711–1720. [Google Scholar] [CrossRef]
- Skinner, R.; Trujillo, A.; Ma, X.; Beierle, E.A. Ketone bodies inhibit the viability of human neuroblastoma cells. J. Pediatr. Surg. 2009, 44, 212–216. [Google Scholar] [CrossRef]
- Tisdale, M.J.; Brennan, R.A. Loss of acetoacetate coenzyme A transferase activity in tumours of peripheral tissues. Br. J. Cancer 1983, 47, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Germain, N.; Dhayer, M.; Boileau, M.; Fovez, Q.; Kluza, J.; Marchetti, P. Lipid Metabolism and Resistance to Anticancer Treatment. Biology 2020, 9, 474–495. [Google Scholar] [CrossRef]
- Wang, J.; Xiang, H.; Lu, Y.; Wu, T.; Ji, G. The role and therapeutic implication of CPTs in fatty acid oxidation and cancers progression. Am. J. Cancer Res. 2021, 11, 2477–2494. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8263643 (accessed on 14 June 2022).
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Cindric, M.; Jakovcevic, A.; Zarkovic, K.; Zarkovic, N. Lipid peroxidation in brain tumors. Neurochem. Int. 2021, 149, 105118. [Google Scholar] [CrossRef] [PubMed]
- McBean, G.J. The transsulfuration pathway: A source of cysteine for glutathione in astrocytes. Amino Acids 2012, 42, 199–205. [Google Scholar] [CrossRef]
- Ogunrinu, T.A.; Sontheimer, H. Hypoxia increases the dependence of glioma cells on glutathione. J. Biol. Chem. 2010, 285, 37716–37724. [Google Scholar] [CrossRef]
- Cliffe, E.E.; Waley, S.G. Acidic peptides of the lens. 6. Metabolism of gamma-glutamyl peptides in subcellular fractions of rabbit liver. Biochem. J. 1961, 79, 118–128. [Google Scholar] [CrossRef]
- Servillo, L.; Castaldo, D.; Giovane, A.; Casale, R.; D’Onofrio, N.; Cautela, D.; Balestrieri, M.L. Ophthalmic acid is a marker of oxidative stress in plants as in animals. Biochim. Biophys. Acta. Gen. Subj. 2018, 1862, 991–998. [Google Scholar] [CrossRef]
- Waley, S.G. Acidic peptides of the lens. 3. The structure of ophthalmic acid. Biochem. J. 1958, 68, 189–192. [Google Scholar] [CrossRef]
- Kombu, R.S.; Zhang, G.F.; Abbas, R.; Mieyal, J.J.; Anderson, V.E.; Kelleher, J.K.; Sanabria, J.R.; Brunengraber, H. Dynamics of glutathione and ophthalmate traced with 2H-enriched body water in rats and humans. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E260–E269. [Google Scholar] [CrossRef] [PubMed]
- Abbas, R.; Kombu, R.S.; Ibarra, R.A.; Goyal, K.K.; Brunengraber, H.; Sanabria, J.R. The dynamics of glutathione species and ophthalmate concentrations in plasma from the VX2 rabbit model of secondary liver tumors. HPB Surg. 2011, 2011, 709052. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Packer, L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996, 10, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.T.; Bao, J.; Furuichi, M.; Yamane, Y.; Gomi, A.; Savaraj, N.; Masuzawa, T.; Ishikawa, T. Frequent coexpression of MRP/GS-X pump and gamma-glutamylcysteine synthetase mRNA in drug-resistant cells, untreated tumor cells, and normal mouse tissues. Biochem. Pharmacol. 1998, 55, 605–615. [Google Scholar] [CrossRef]
- McLellan, L.I.; Wolf, C.R. Glutathione and glutathione-dependent enzymes in cancer drug resistance. Drug Resist. Updat. 1999, 2, 153–164. [Google Scholar] [CrossRef]
- Ogretmen, B.; Bahadori, H.R.; McCauley, M.D.; Boylan, A.; Green, M.R.; Safa, A.R. Co-ordinated over-expression of the MRP and gamma-glutamylcysteine synthetase genes, but not MDR1, correlates with doxorubicin resistance in human malignant mesothelioma cell lines. Int. J. Cancer 1998, 75, 757–761. [Google Scholar] [CrossRef]
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, L.J.Y.; Prager, B.C.; Flavahan, W.A.; Liu, X.; et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat. Neurosci. 2017, 20, 661–673. [Google Scholar] [CrossRef]
- Zhou, W.; Wahl, D.R. Purine metabolism promotes radioresistance and is a therapeutic target in glioblastoma. Mol. Cell Oncol. 2020, 7, 1834902. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliva, C.R.; Ali, M.Y.; Flor, S.; Griguer, C.E. Effect of Expression of Nuclear-Encoded Cytochrome C Oxidase Subunit 4 Isoforms on Metabolic Profiles of Glioma Cells. Metabolites 2022, 12, 748. https://doi.org/10.3390/metabo12080748
Oliva CR, Ali MY, Flor S, Griguer CE. Effect of Expression of Nuclear-Encoded Cytochrome C Oxidase Subunit 4 Isoforms on Metabolic Profiles of Glioma Cells. Metabolites. 2022; 12(8):748. https://doi.org/10.3390/metabo12080748
Chicago/Turabian StyleOliva, Claudia R., Md Yousuf Ali, Susanne Flor, and Corinne E. Griguer. 2022. "Effect of Expression of Nuclear-Encoded Cytochrome C Oxidase Subunit 4 Isoforms on Metabolic Profiles of Glioma Cells" Metabolites 12, no. 8: 748. https://doi.org/10.3390/metabo12080748
APA StyleOliva, C. R., Ali, M. Y., Flor, S., & Griguer, C. E. (2022). Effect of Expression of Nuclear-Encoded Cytochrome C Oxidase Subunit 4 Isoforms on Metabolic Profiles of Glioma Cells. Metabolites, 12(8), 748. https://doi.org/10.3390/metabo12080748